Aysen Orman1, Nilay Hakan2, Atika Caglar1, Mustafa Aydin1, Erdal Taskin1.
1Department of Pediatrics-Neonatology, Firat University School of Medicine, Elazig, Turkey,
2Department of Pediatrics-Neonatology, Sitki Kocman University School of Medicine, Mugla, Turkey.
ADDRESS FOR CORRESPONDENCE
Mustafa AYDIN, MD, Department of Pediatrics-Neonatology, Firat University School of Medicine, 23119, Elazig / Turkey.
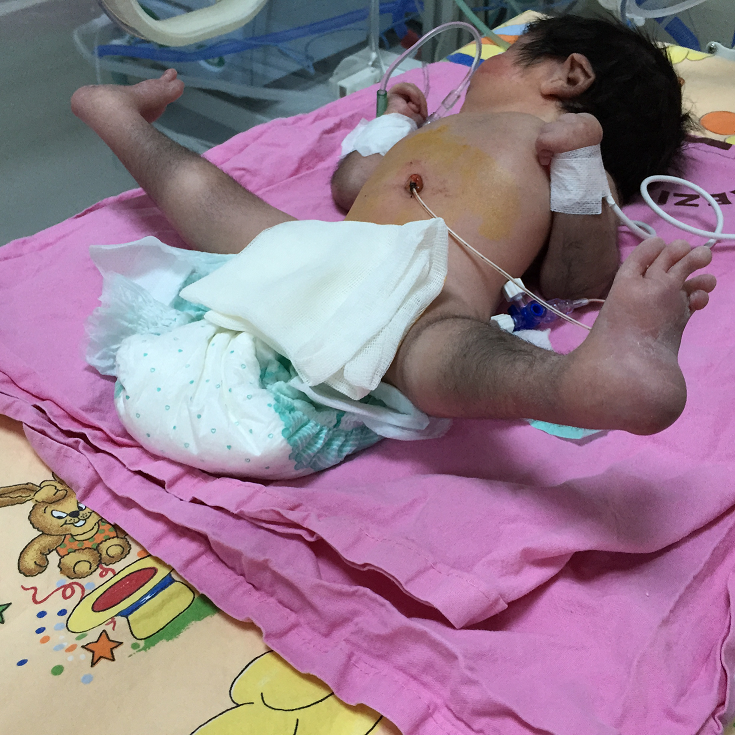
Email: dr1mustafa@hotmail.com | | Keywords | | Deletion, chromosome 10p12.1, anomaly, neonate | | | A 2-day-old newborn was referred to our neonatal intensive care unit (NICU) because of multiple congenital malformations accompanied by hypertrichosis. She was born via vaginal delivery at 38 weeks of gestation from a 27-year-old multipara woman as the second child. Elder sister was healthy. Antenatal history with respect to maternal drug use, exposure to radiation, teratogens, or mutagenic chemicals was unremarkable. Likewise, family history was also unremarkable. Parents were second-degree cousins. The birth weight was 2450 g (3-10th centile), head circumference 32 cm (<3rd centile). Physical examination showed craniofacial malformations including microcephaly, abnormally shaped skull, down slanted palpebral fissures, epicanthal folds, hypertelorism, low nasal bridge, micrognathia, dysmorphic low set ears, and short neck. There was camptodactyly in both hands, distinct anomalies in toes, bird foot, and bilateral stick legs. Both hip joints were in flexion and external rotation posture, both patellae were located posteriorly. The patient had diffuse hypertrichosis with prominent dark hair over, especially on the legs, arms, and back (Figure 1). A systolodiastolic murmur with a grade of 2/6 was heard in the pulmonary area. Karyotype analysis yielded the 46,XY,del(10)(p12.1). Echocardiographic examination revealed patent ductus arteriosus (PDA). Brain magnetic resonance imaging showed no abnormality. Complete blood count and biochemical tests were all within the normal limits. Her clinical follow-up manifested the motor and mental developmental delay, and unilateral sensorineural hearing loss by auditory brainstem response. After that, she died on the 8th month of life.
Figure 1. Distinct dysmorphic features of feet and toes with hypertrichosis
A deletion from any of the p11 or p12 bands is called a proximal deletion. So, deletion of the proximal short arm of chromosome 10 represents a rare genetic alteration, especially those encompassing the chromosomal region 10p11 and 10p12.1 Deletion at chromosome 10p12-p11 has overlapping phenotypes with DeSanto-Shinawi syndrome (DESSH) which is caused by heterozygous mutation in the WAC gene on chromosome 10p11. DESSH is a rare neurodevelopmental disorder characterized by global developmental delay apparent in infancy or early childhood and associated with characteristic dysmorphic facial features, gastrointestinal and mild ocular abnormalities, as well as behavioral problems. The phenotypic expression of deletions of the chromosome 10p are variable, but the craniofacial malformations constitute a more consistent finding, as seen in the present patient. Those cases with the deletions of chromosome 10p.12 have also heterogeneous extremity anomalies including syndactyly, preaxial polydactyly, club hand and feet, clinodactyly and hypoplastic distal phalanges.1,2 Likewise, our patient had distinct lower limb anomalies. Half of the patients had congenital heart diseases which are the major cause of death within the first months of life. However, our patient did not have any cardiac pathology except PDA.
Infants and children with 10p12 deletion have atypical behavior pattern, learning disability, cognitive difficulties, hearing loss, and feeding difficulties.3 Though, cranial imaging was normal in our case, deletions of the genes involved in the formation of the forebrain involving 10p12.1 result in holoprosencephaly. Besides, some patients had arhinencephaly (absence of olfactory bulbs without defects of brain cleavage), which may be considered as a mild manifestation of the same defect.2 Therefore, this part of 10p12.1 has to contain a gene involved in the formation of the forebrain. These pathologies could be the causes of psychomotor delay and hearing disability, as seen in the present case. In addition, hypertrichosis was present in our case which previously rarely described.
In conclusion, we report a rare case of a newborn infant with a deletion of 10p12.1 that was characterized by dysmorphic craniofacial features, hypertrichosis, limb anomalies, and developmental delay, and hearing impairment.
| | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Abdelhedi F, El Khattabi L, Essid N, Viot G, Letessier D, Lebbar A, Dupont JM. A de novo 10p11.23-p12.1 deletion recapitulates the phenotype observed in WAC mutations and strengthens the role of WAC in intellectual disability and behavior disorders. Am J Med Genet A 2016; 170:1912-7. [CrossRef] [PubMed]
- Wentzel C, Rajcan-Separovic E, Ruivenkamp CA, Chantot-Bastaraud S, Metay C, Andrieux, J et al. Genomic and clinical characteristics of six patients with partially overlapping interstitial deletions at 10p12p11. Eur J Hum Genet 2011; 19:959-64. [CrossRef] [PubMed] [PMC free article]
- Unique. Understanding chromosome disorders. 10p deletions. Available at URL: https://www.rarechromo.org/media/information/Chromosome%2010/10p%20deletions%20FTNW.pdf. Accessed on 14th September 2019
DOI: https://doi.org/10.7199/ped.oncall.2020.17
|
| Cite this article as: | | Orman A, Hakan N, Caglar A, Aydin M, Taskin E. Hypertrichosis in a Newborn with Deletion of the Short Arm of Chromosome 10 (10p12.1). Pediatr Oncall J. 2020;17: 61-62. doi: 10.7199/ped.oncall.2020.17 |
|