Nibedita Mitra, N Kannan.
Department of Pediatrics, Southern Railway Head Quarter Hospital, Perambur, Chennai, India.
ADDRESS FOR CORRESPONDENCE
Dr. Nibedita Mitra, 431/C, Brahmaputra Apartment, Pilkington Road, Ayanavaram, Chennai 600023, India.
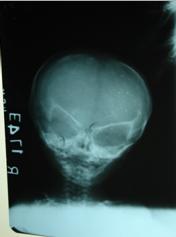
Email: nibeditamitra1@yahoo.co.in | | Keywords | | natal teeth, hypotrichosis, hypolastic mandible, skin atrophy | | | | Keywords | A twenty-day-old neonate was referred to our hospital for feeding difficulty and noisy respiration. She is the first-born child to parents of nonconsanguinous marriage, delivered after two years of marriage. Baby was delivered at term after an uneventful pregnancy with a birth weight of 2.2kg, head circumference was 33 cms and length was 45 cms. Mother is twenty-five years old, and father's age is twenty-nine years. The first product of conception was aborted at two months gestation. On examination, the neonate had two natal teeth (upper central incisors), severe micrognathia, a small mouth and high arched palate and a small and thin tongue. There was frontal prominence with wide-open posterior and anterior fontanel and widely open sutures. The nose was small and beaked. Scattered patches of thin hypo pigmented skin were present over face, trunk, back and limbs. The scalp hair was sparse and thin with areas of complete baldness mainly along the lambdoid suture. Eyebrows were sparse but eyelashes were normal. There was microphthamia but no congenital cataract (Figure 1). The nails, ears, external genitalia and limbs were normal. Skull radiograph shows hypoplastic mandible (Figure 2). Ultrasonography of cranium, abdomen and pelvis did not reveal any anomaly Uterus size was within normal limits for the age. Based on the above clinical presentation a diagnosis of Hallerman-Streiff Syndrome was made. The baby is not able to suck at the breast but is accepting feeds well with a palaade. She has been evaluated by an ophthalmologist and has been advised regular follow up. Proper positioning of the baby has been advised to the mother.
Figure 1: Natal teeth, parrot beaked nose, micrognathia, atrophic skin, hypotrichosis.
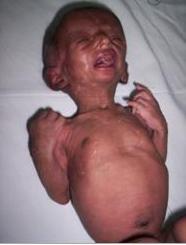
Figure 2: X-ray showing hypoplastic mandible
Hallermann-Streiff syndrome occurs sporadically and is not associated with chromosomal anomalies. It affects both male and female in all ethnic groups (1, 2). It is a rare genetic condition and is also known as Francois dyscephaly syndrome (3). Dyscephaly can be defined as malformation of the cranium and bones of the face. Until 1981, around 150 cases have been reported. Since then another 10 cases have been reported. Hallermann-Streiff syndrome consists of abnormalities of the skull, malformation of the facial skeleton and jaw, dental anomalies including the presence of erupted teeth at birth, localized hypotrichosis, congenital abnormalities of the eyes, dwarfism and motor and mental retardation which is occasionally present. (1,2) The skull is brachycephalic in most instances, with a discrepancy in size of large cranial vault and small facial skeleton. The head circumference may be normal or lower limit of normal. Prominent frontal and parietal eminences are present. The posterior fontanel is frequently open at birth and remains so for a long time. The sutures are usually widely open. The mouth is small and the palate is high arched. The most striking feature of facial skeleton is the hypoplastic mandible. The nose is small and beaked with a sharp nasofrontal angle and often a deviated nasal septum. The narrow upper air passage lead to feeding difficulty in the newborn period and early infancy and minor respiratory infection in later life can cause airway obstruction. Dental anomalies include presence of teeth at birth and presence of extra teeth. Underdevelopment of tooth enamel and cavities are also common. The striking feature is the diminished hair growth on the scalp. The hair is thin and sparse with areas of complete baldness, the latter either consists of patches of alopecia along the coronal and lambdoid sutures or more typically there is complete baldness over the frontal area with anterior border at the level of coronal sutures. The most common anomalies of the eyes are bilateral microphthalmia, congenital cataract, ocular nystagmus and strabismus. In several of the reported cases spontaneous resorption of the cataract occurred. Cataract has been reported in 26 cases and chorioretinal atrophy has been reported in three cases (4). Bodily growth is severely retarded in most cases resulting in proportionate dwarfism with retarded bone age. Mental impairment has been reported in some cases. There is no cure for Hallerman-Streiff syndrome. Affected individuals need a team of specialized doctors for treating the various problems, which can occur. Assessment by a dentist, dental surgeon and orofacial surgeon may be required to evaluate the teeth and difficulties caused by small chin and mouth. Individuals affected with Hallermann-Streiff syndrome may have normal intelligence and life span when complications of this disorder are properly managed (5). | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Hallerman W. Vogelgesult un cataractous congenita. Klin Monatsbl Augenheilkd. 1948; 113: 315.
- Streiff HB. Dvsmorphic mandibulo-faciale (tete d' oiseaiu) et alterations oculaires. Ophthalmologica. 1950; 120: 79. [CrossRef]
- Franscois J, Troncoso VV. Francois dyscephalic syndrome and skin manifestations. Ophthalmologica. 1981; 183: 63-67. [CrossRef]
- Neki AS. Hallermann-Streiff syndrome. Indian J Ophthalmol 1993; 41: 83-84. [PubMed]
- Hoefnagel D, Benirschke K. Dyscephalia Mandibulo-oculo-facialis*(Hallermann-Streiff Syndrome) Arch Dis Child. 1965; 40: 57-61. [CrossRef]
|
| Cite this article as: | | Mitra N, Kannan N. Hallermann-Streiff Syndrome. Pediatr Oncall J. 2011;8: 48-49. |
|