Megha Popat, Vedant Popat.
Department of Pediatrics, Om Babycare Hospital, Near R-world cinema, Besides Pradhyumannagar police station, Rajkot, Gujarat, India.
ADDRESS FOR CORRESPONDENCE
Vedant Popat, Om Babycare Hospital, Near R-world cinema, Besides Pradhyumannagar police station, Rajkot, Gujarat - 360001, India.
Email: vedant1210@gmail.com | | Abstract | Background: Copy number variations result in diverse clinical phenotypes, often presenting with dysmorphism, neurodevelopmental impairment and growth abnormalities.
Clinical Description: Full term male baby presented at 1.5 months with failure to thrive. On examination, baby had dysmorphic facies, microcephaly, cholestasis and chordee. During hospitalization, baby had episodes of asymptomatic hypoglycemia and seizures. Feeding difficulties were noted with baby not gaining adequate weight despite optimal nutrition. Genetic evaluation revealed partial deletion of chromosome 7q and duplication of chromosome 12q.
Conclusion: This case highlights various genotype-phenotype correlations in chromosomal disorders. Early diagnosis is crucial for management, prognostication, genetic counseling and future reproductive decision-making along with multidisciplinary follow up. | | | | Keywords | | Failure to thrive, Cholestasis, Encephalopathy, Syndromic, Whole exome. | | | | Introduction | Chromosomal abnormalities are a significant cause of congenital anomalies, dysmorphic features and neurodevelopmental disorders in the infancy period. 1
Deletions involving the long arm of chromosome 7 have been associated with growth restriction, microcephaly, craniofacial dysmorphism and developmental delay, while duplications of chromosome 12q are linked to neurological impairment, congenital anomalies and various dysmorphic features. Individually, these chromosomal abnormalities are rare; however, the coexistence of both 7q deletion and 12q duplication in a single patient is exceedingly uncommon.
Clinical Description
Full term 38 weeks male baby was born via lower segment caesarean section (LSCS) to a G3P2A1L1 mother via non-consanguineous marriage with a birth weight of 2620 gm (~10th centile), length of 50 cm (~50th centile) and head circumference of 31 cm (<3rd centile). Baby cried immediately after birth and was shifted with mother for routine care.
Antenatal period was normal with no significant history of any maternal illness, drug ingestion or radiation exposure. There was history of a spontaneous abortion at 3 months of age during the first pregnancy but no further evaluation was done. Antenatal scans in each trimester were normal.
Baby presented at 1.5 months of age with complaint of feed intolerance and not gaining weight. On admission, baby had weight gain of only 140 grams in 1.5 months. On admission, baby had fair activity with moderate level of spontaneous movements and responsiveness. Baby had reduced vigor with a less forceful cry. Baby was vitally stable on room air. Icterus was present upto legs.
On physical examination, there was upslanting of both eyes with large ears (Figure 1). Baby had microcephaly with an occipito-frontal circumference of 31 cm (<3rd centile according to Intergrowth-21 chart). There was no organomegaly present. Genitals were normal but there was downward curvature of penis suggestive of chordee. The baby had poor acceptance of breastfeeding (BF). This feeding difficulty was also reflective of ineffective suck-swallow coordination or underlying neurological or metabolic compromise.
Baseline blood investigations were sent along with ABG (Table 1) which were suggestive of conjugated hyperbilirubinemia indicative of neonatal cholestasis. During the hospital course, baby was observed to have multiple episodes of asymptomatic hypoglycemia. Enteral feeds were managed with optimal calorie intake. Evaluation for a metabolic cause was also done (Table 1). TORCH profile was sent to rule out congenital infections which turned out negative. Screening for other abnormalities was done (Table 2). Ophthalmology screening was also done suggestive of normal retina, fundus and anterior segment.
Baby was started on high calorie feeds with multivitamin supplements and ursodeoxycholic acid. At 2 months of age, subtle seizures were recognized in form of cyclic movements of both upper and lower limbs with repetitive kicking. EEG and MRI Brain were done. Syrup levetiracetam was started along with tablet pyridoxine.
Presence of abnormal facial morphology, feeding difficulties and seizure activity, raised suspicion for syndromic disorder and hence whole exome sequencing (WES) was sent (Table 3).
Baby was discharged on 66th day of life with a weight of 3300 grams (<3rd centile), length of 60 cm (50-75th centile) and head circumference of 34 cm (<3rd centile). Relatives were taught proper method of breast feeding and spoon feeding with supplemental multivitamins along with limb and oromotor physiotherapy.
Serial follow up visits were planned to assess weight gain, head circumference, feeding adequacy and developmental milestones. Screening for associated comorbidities was also advised.
Parents were counseled regarding possible neurological implications, need for long-term follow up and involvement of a multidisciplinary team for optimal care. Parental karyotyping was also advised for subsequent pregnancies.
Figure 1. Baby with syndromic facies in form of upslanting eyes, large ears, microcephaly, conjugated hyperbilirubinemia.
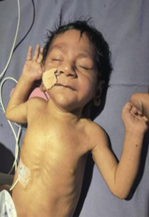
Table 1. Laboratory Investigations.
| |
DOL-30 |
DOL-42 |
DOL-48 |
DOL-53 |
DOL-60 |
DOL-66 |
| Hemoglobin (g/dl) |
10.9 |
|
|
10.6 |
|
10.8 |
| Total leukocyte count (pcmm) |
11490 |
|
|
11400 |
|
13850 |
| Platelets (pµl) |
620000 |
|
|
615000 |
|
626000 |
| CRP (mg/l) |
0.5 |
8.3 |
2.6 |
2.0 |
|
|
| Creatinine (mg%) |
0.44 |
|
|
|
|
0.36 |
| Bilirubin (mg/dl) (Total / Direct / Indirect) |
12.52 / 8.27 / 4.25 |
8.59 / 6.73 / 1.86 |
9.11 / 7.12 / 1.99 |
7.3 / 5.43 / 1.87 |
4.21 /2.83 / 1.38 |
1.66 / 1.41 / 0.25 |
| Alkaline phosphatase (U/L) |
|
612 |
|
572 |
|
453 |
| SGPT (U/L) |
|
228 |
197 |
166 |
|
94 |
| SGOT (U/L) |
|
277 |
225 |
157 |
|
69 |
| Ammonia (mcg/dl) / Lactate (mmol/l) |
220 / 3.25 |
84 |
74 |
|
|
59 |
| Ionized Calcium (mmol/l) |
1.49 |
1.31 |
|
|
|
1.28 |
| PT / INR |
18 / 1.44 |
|
|
|
|
14 / 1.21 |
Table 2. Screening tests and other advanced tests.
| Investigation |
Result |
| Cytomegalovirus IgM |
Negative |
| TORCH |
Negative |
| Tandem mass spectrometry |
Negative |
| USG Cranium & Abdomen |
Normal |
| 2d Echo |
Tiny slit-like apical muscular VSD with L ? R flow (hemodynamically insignificant) |
| OAE |
Normal |
| Xray Spine |
Normal |
| MRI Brain |
Normal |
| EEG |
Multifocal and generalized epileptiform discharges with presence of burst attenuation patterns ? s/o evolving early epileptic encephalopathy |
| Whole exome sequencing |
• Heterozygous pathogenic variant ? partial deletion of chromosome 7q34-36.3 region (involving 116 genes)
• Variant of uncertain significance (VUS) ? partial duplication of chromosome 12q24.31-24.33 region (involving 65 genes). |
Table 3. Primary findings of Whole exome sequence report.
| Genomic Region |
Variation |
Zygosity |
Disease |
Inheritance |
Variant classification |
| chr7q34-36.3 |
chr7:(?142560955)(158937493_?)del(chr7q34-36.3 partial deletion) |
Heterozygous |
Chromosome 7q deletion syndrome |
Dominant |
Pathogenic |
| chr12q24.31-24.33 |
chr12:(?123380468)(133810972_?) [3](chr12q24.31-24.33 partial duplication) |
NA |
Chromosome 12q duplication syndrome |
NA |
Variant of Uncertain Significance (VUS) |
| | | | Discussion | Current case, with partial deletion of chromosome 7q and duplication of chromosome12q, illustrates a unique genomic imbalance contributing to multisystem involvement. Such combined rearrangements significantly influence the phenotypic severity2,3 resulting in a more severe phenotype.4
Differentials such as Smith-Lemli-Opitz syndrome (SLOS), Kabuki syndrome and chromosome 1p36 deletion were kept as they present with similar overlapping features.
Few cases of isolated chromosomal deletions or duplications have been identified but combination has been rare. Present case did not demonstrate features of holoprosencephaly, such as midline facial defects or structural brain abnormalities on neuroimaging which suggested that the identified chromosomal imbalance did not involve critical regions associated with forebrain cleavage defects.5,6 Vassilis et.al7, demonstrated correlation between genomic disruptions and holoprosencephaly in an adult patient, while our case exhibited a distinct phenotype despite the presence of significant chromosomal rearrangement. | | | | Conclusion | | This case highlights the importance of considering rare chromosomal rearrangements in neonates presenting with multiple congenital anomalies and neurological manifestations. Early diagnosis using advanced genomic techniques such as chromosomal microarray or whole exome sequencing is crucial for accurate diagnosis, prognostication and genetic counseling, particularly in guiding recurrence risk and future pregnancy planning. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. The American Journal of Human Genetics. 2010 May 14;86(5):749-64. [CrossRef] [PubMed] [PMC free article]
- Schinzel A. Catalogue of unbalanced chromosome aberrations in man. Walter de Gruyter GmbH & Co KG; 2020 Oct 26.
- Menten B, Maas N, Thienpont B, Buysse K, Vandesompele J, Melotte C, de Ravel T, Van Vooren S, Balikova I, Backx L, Janssens S. Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: a new series of 140 patients and review of published reports. Journal of medical genetics. 2006 Aug 1;43(8):625-33. [CrossRef] [PubMed] [PMC free article]
- McKusick VA. Mendelian Inheritance in Man and its online version, OMIM. The American Journal of Human Genetics. 2007 Apr 1;80(4):588-604. [CrossRef] [PubMed] [PMC free article]
- Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphanet journal of rare diseases. 2007 Feb 2;2(1):8. [CrossRef] [PubMed] [PMC free article]
- Solomon BD, Mercier S, Vélez JI, Pineda‐Alvarez DE, Wyllie A, Zhou N, Dubourg C, David V, Odent S, Roessler E, Muenke M. Analysis of genotype-phenotype correlations in human holoprosencephaly. InAmerican Journal of Medical Genetics Part C: Seminars in Medical Genetics 2010 Feb 15 (Vol. 154, No. 1, pp. 133-141). Hoboken: Wiley Subscription Services, Inc., A Wiley Company. [CrossRef] [PubMed] [PMC free article]
- Paspaliaris V, Vrachnis N, Iliodromiti Z, Antonakopoulos N, Papaioannou G, Vlachadis N, Anastasiadou F, Sotiriou S, Garas A, Thomaidis L, Manolakos E. 7q deletion/12q duplication is the possible cause of an alobar holoprosencephaly case. Molecular Syndromology. 2017 Dec 20;9(1):52-7. [CrossRef] [PubMed] [PMC free article]
DOI: https://doi.org/10.7199/ped.oncall.2027.58
|
| Cite this article as: | | Popat V, Popat M. A rare presentation of syndromic neonate presenting with cholestasis and neurodevelopmental impairment revealing complex chromosomal rearrangement. Pediatr Oncall J. 2026 Jun 29. doi: 10.7199/ped.oncall.2027.58 |
|