Rhea Shriyan, Pradeep Debata, Anita Yadav, Nidhi Chopra.
Department of Pediatrics, Vardhman Mahavir Medical College & Safdarjung Hospital, Delhi, India.
ADDRESS FOR CORRESPONDENCE
Dr Pradeep Debata, Associate Professor, Department of Pediatrics, VMMC & Safdarjung Hospital, Delhi, India.
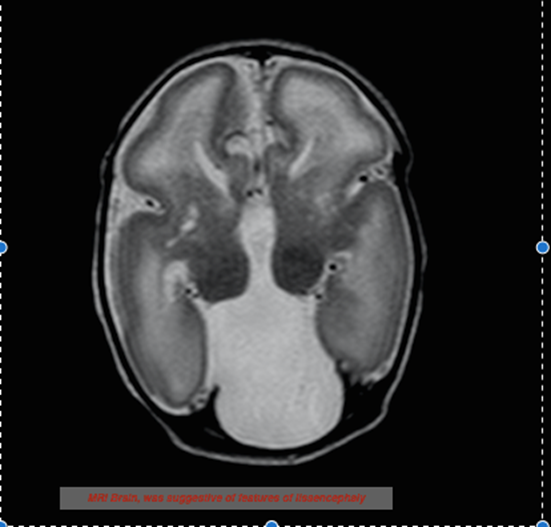
Email: drpkdebata@gmail.com | | Abstract | | X-linked lissencephaly with corpus callosum agenesis and ambiguous genitalia (XLAG) is a newly recognised rare syndrome of severe neurological onset, caused by mutation in the aristaless-related homeobox (ARX) gene (Xp22.13). A term baby had abnormal genitalia with multiple multifocal clonic seizures, starting at 2 hours of life whose MRI of Brain revealed lissencephaly with corpus callosum agenesis suggestive of XLAG. | | | | Introduction | | Neuronal migrational disorders are an important cause of intractable epilepsy of neonatal onset. X-linked lissencephaly, absent corpus callosum with ambiguous genitalia (XLAG) is a rare and recently reported syndrome. The first case was described by Dobyns et.al in 1999. (1) Thereafter there are only 15 reported cases in literature. (2) It is caused by a mutation of the aristaless-related homeobox (ARX) homeobox gene (Xp22.13), which is situated around the ventricles, neocortex, hippocampus and also in the testis and pancreas. (3) Herein, we present a neonate with features consistent with this syndrome. | | | | Case Report | A term neonate, appropriate for gestational age, was born by normal vaginal delivery to a third gravida mother at 39+2 weeks of gestation. There was no history of consanguinity. The baby cried immediately after birth with an APGAR score of 8,and 9 at 1 and 5 minutes respectively. Birth weight was 3 kg. A previous male sibling died on Day 10 with seizures that started at 1 hour of life. Another female sibling aged 10 years was healthy. On examination, the current child had a head circumference of 33 cm. Ambiguous genitalia was noted in the form of micropenis (stretched penile length 1.2 cm), hypospadiasis, fused labioscrotal folds and bilaterally impalpable gonads. He had a flat occiput, lethargy, axial hypotonia, poor suck and partial Moro’s reflex. At 2 hours of life, the baby had the first episode of multifocal clonic seizure, which was aborted by phenobarbitone. Thereafter, there were multiple seizure episodes which required phenytoin and midazolam infusion for control. Intermittent episodes of hypothermia and diarrhea were observed during hospital stay. Complete blood counts, serum electrolytes and calcium, renal and liver function tests were normal. Hypoglycemia was not documented during any seizure. Sepsis workup was negative and cultures was sterile. Arterial blood gas analysis revealed, pH: 7.37, pCO2: 40 mmHg, pO2: 86 mmHg, HCO3 – 22.6 mEq/L, lactate: 1.2 mmol/L. Blood ammonia was 56 mol/L. Tandem mass spectrophotometry and gas chromatography-mass spectrophotometry were normal. Ultrasound (USG) skull revealed absence of corpus callosum and presence of dorsal interhemispheric cyst, communicating with the third ventricle. MRI of Brain showed lissencephaly. In addition to the above findings, there were smooth surface with lack of gyral pattern diffusely involving all cerebral hemispheres, bilaterally narrow sylvian fissure and mildly enlarged ventricles (Figure 1). EEG was suggestive of multifocal epileptiform discharges. Testicular/mullerian structures could not be identified on USG of pelvis. MRI of abdomen and pelvis showed a small well defined structure of hypo-isoechoeic on T1W and T2W imaging in the left inguinal region suggestive of testis. Karyotyping confirmed 46,XY genotype. Genetic analysis was planned but could not be performed due to financial constraints of the parents. However, the nature of the disease, prognosis and possibility of recurrence in subsequent pregnancies was clearly explained. The baby was managed with intravenous fluids, multiple antiepileptics and supportive care.
Figure 1: MRI brain showing lissencephaly and absent corpus callosum
| | | | Discussion | Neonatal seizures are a common pediatric emergency, with a myriad of causes ranging from hypoxic ischemic encephalopathy, intracranial haemorrhage, infections, metabolic to congenital brain malformations. (4) In our patients, there was no birth asphyxia, intracranial bleed and the metabolic profile and sepsis workup were normal. Therefore, in the presence of ambiguous genitalia with refractory seizures investigations were targeted at studying any underlying brain malformation. Brain malformations account for 5-10% of neonatal seizures. Among these, lissencephaly is rare neuronal migration disorder, with X-linked mode of inheritance being described only recently. (5) Similarly our patient was a male and the other sibling who died of seizures was also a male while the female sibling was healthy. Our patient had multifocal clonic seizures, however, myoclonic and generalised tonic clonic seizures have also been reported. (6,7) In addition, lethargy and hypotonia were observed on initial neurological examination similar to other studies. (2,6) Neuroimaging in few of the reported cases of XLAG suggest a posterior to anterior severity gradient with posterior agyria and anterior pachygyria. (1,2,6) In our patient too, there was diffuse loss of gyral pattern with ventriculomegaly and absent corpus callosum. Genital abnormalities noted in our patient included microphallus, hypospadiasis, fused labioscrotal folds and bilaterally impalpable gonads. Minocha et al and many others reported similar presentations. (1,2,6,8) Spinosa et al (9) and Okazaki et al (10) have reported the occurrence of diarrhea similar to our patient though the mechanism of pathogenesis is not well understood. Hypothalamic dysfunction in the form of temperature instability was seen in our patient as reported previously. (1,9)
ARX gene mutations may manifest as XLAG, X-linked infantile spasms (West syndrome), X-linked myoclonic epilepsy with spasticity and mental retardation, X-linked mental retardation, Partington syndrome (mental retardation, dystonic movements of the hands, and dysarthria), Proud syndrome (acquired microcephaly, mental retardation, agenesis of the corpus callosum, and characteristic facies), or hydranencephaly with ambiguous genitalia. (8,9) | | | | Conclusion | | X-linked lissencephaly, absent corpus callosum with ambiguous genitalia (XLAG) should be considered in male infants with refractory seizures with ambiguous genitalia. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Dobyns WB, Berry-Kravis E, Havernick NJ, Holden KR, Viskochil D. X-linked lissencephaly with absent corpus callosum and ambiguous genitalia. Am J Med Genet. 1999;86:331–7. [CrossRef]
- Motlagh AJ, Zahedpasha Y, Ahmadpourkacho M. X-Linked Lissencephaly with absent corpus callosum and ambiguous genitalia: A case report. Iran J Neonatol. 2016;7:75-9.
- Miura H, Yanazawa M, Kato K, Kitamura K. Expression of a novel aristaless related homeobox gene ‘Arx' in the vertebrate telencephalon, diencephalon and floor plate. Mech Dev. 1997; 65:99-109. [CrossRef]
- Prasad M, Chow G. Neonatal seizure: What is the cause? BMJ. 2012;345:e6003. [CrossRef] [PubMed]
- Panda S, Tripathi M, Jain S, Sharma P. X-linked lissencephaly in an Indian family. Neurol India 2003;51:392-3. [PubMed]
- Bonneau D, Toutain A, Laquerrière A, Marret S, Saugier‐Veber P, Barthez MA, et al. X‐linked lissencephaly with absent corpus callosum and ambiguous genitalia (XLAG): clinical, magnetic resonance imaging, and neuropathological findings. Ann Neurol. 2002; 51(3):340-9. [CrossRef] [PubMed]
- Uyanik G, Aigner L, Martin P, Gross C, Neumann D, MarschnerSchäfer H, et al. ARX mutations in X-linked lissencephaly with abnormal genitalia. Neurology. 2003;61:232-5. [CrossRef] [PubMed]
- Minocha P, Choudhary A, Shambhavi, Sitaraman S. A Neonate with X-linked Lissencephaly with Ambiguous Genitalia. J Pediatr Neurosci. 2017; 12: 80–82. [CrossRef] [PubMed] [PMC free article]
- Spinosa MJ, Liberalesso PB, Vieira SC, Olmos AS, Löhr A Jr. Lissencephaly, abnormal genitalia and refractory epilepsy: Case report of XLAG syndrome. Arq Neuropsiquiatr. 2006;64:1023-6. [CrossRef] [PubMed]
- Okazaki S, Ohsawa M, Kuki I, Kawasaki H, Koriyama T, Ri S, et al. Aristaless-related homeobox gene disruption leads to abnormal distribution of GABAergic interneurons in human neocortex: Evidence based on a case of X-linked lissencephaly with abnormal genitalia (XLAG). Acta Neuropathol. 2008;116:453-62. [CrossRef] [PubMed]
DOI: https://doi.org/10.7199/ped.oncall.2018.31
|
| Cite this article as: | | Shriyan R, Debata P, Yadav A, Chopra N. X-Linked Lissencephaly and Ambiguous Genitalia. Pediatr Oncall J. 2018;15: 75-76. doi: 10.7199/ped.oncall.2018.31 |
|