Krishna Chaitanya1, Archana Addanki1, Nisha Deshpande2, Rajesh Badani3.
1Department of Pediatrics, Aditya Birla Memorial Hospital, Pune, India,
2Department of Pediatric Neurology, Aditya Birla Memorial Hospital, Pune, India,
3Department of Pediatric Cardiology, Aditya Birla Memorial Hospital, Pune, India.
ADDRESS FOR CORRESPONDENCE
Department of Pediatrics, Aditya Birla Memorial Hospital, Chinchwad, Pune, Maharashtra, India 411033.
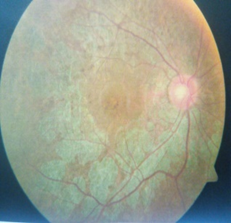
Email: dkrishnac7@gmail.com | | Abstract | | Kearns-Sayre syndrome is a rare mitochondrial deletion syndrome characterized by triad of cardiac conduction defects, chronic progressive external ophthalmoplegia and pigmentary retinopathy. We present a 15 years old child who was diagnosed and treated as myasthenia gravis for several years and currently presented with complete heart block. He was finally diagnosed as having KSS on mitochondrial genome sequencing analysis. Thus, KSS and MG are mimics and a high suspicion of KSS should be kept in patients who are diagnosed as MS and additionally have cardiac problems. | | | | Keywords | | KSS; syncope; complete heart block; CPEO; Myasthenia Gravis | | | | Introduction | | Kearns-Sayre syndrome (KSS) is a mitochondriopathy characterized by multiorgan dysfunction, which typically takes place before twenty years of age.1 It was described in 1958 by Thomas Kearns and George Sayre through a report of a case that presented with external ophthalmoplegia, pigmentary retinopathy and cardiac conduction block (CCB), which was later named Kearns-Sayre syndrome.1 Myasthenia gravis (MG) is a long-term neuromuscular disease that leads to varying degrees of skeletal muscle weakness.2 The most commonly affected muscles are those of the eyes, face, and swallowing.2 It can result in double vision, drooping eyelids, trouble talking, and trouble walking.2 MG usually mimics mitochondrial myopathy and Lambert Eaton syndrome. Cardiac manifestations of MG include features of myocarditis, arrhythmias and failure but are very rare.3 We present a 15 years old child who was diagnosed and treated as MG for several years and was finally diagnosed as having KSS on mitochondrial genome sequencing analysis. | | | | Case Report | A 15 years old male presented with progressive loss of vision over the past 2 years associated with bilateral ptosis, generalized fatigue, small stature, delayed pubertal development, dyspnea on moderate effort, palpitations and giddiness and an attack of syncope. He had been diagnosed as MG in view of electromyography (EMG) findings suggestive of decremental response and was treated with pyridostigmine for last few years at a local hospital with no clinical improvement. Physical examination revealed a marked reduction in visual acuity, bilateral external ophthalmoplegia and bilateral ptosis. Cardiovascular monitoring showed irregular heart rhythm. Electrocardiogram (ECG) showed complete heart block (Fig 1). Serum lactate and pyruvate were normal. Serum thyroid stimulating hormone, parathyroid hormone and growth hormone levels were normal. Transthoracic echocardiogram was normal. Fundoscopy showed retinitis pigmentosa (Fig 2). In view of above findings, the possibility of mitochondrial inherited disorder, most probably KSS was considered. Child was hospitalized in intensive care unit and subsequently a permanent pacemaker implantation was done. Mitochondrial genome sequencing analysis of the child was done which showed upto 6.5 kb mitochondrial DNA deletions from the cells collected from his peripheral blood which is suggestive of KSS. He was started on L-Carnitine, coenzyme-Q, calcium and folate.
Figure 1. ECG showing complete heart block
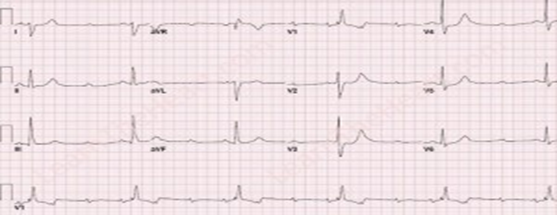
Figure 2. Funduscopy shows retinitis pigmentosa
| | | | Discussion | KSS is a genetic disorder caused by mutations in the mitochondrial DNA. Tissues with high energy demand such as muscle and nervous system are particularly vulnerable to mitochondrial dysfunction, a consequence of deletions, rearrangements or other mutations in mitochondrial DNA.4 Initial presentation of KSS closely mimics that of MG. Ophthalmological, cardiological and endocrinological symptoms present at later stages. Mitochondrial DNA (mt DNA) rearrangements are a key molecular feature of this disease, which manifest a broad phenotypic spectrum. The phenotypic expression depends directly on the number of mutations in alleles and systems affected. Estimated case incidence is 1.6 cases per 100,000 population.5 Disease progresses slowly over years.
KSS is characterized by three main features: a) Typical onset before age of 20 years, although may occur in infancy or adulthood; b) Paralysis of specific eye muscles - chronic progressive external ophthalmoplegia (CPEO); c) Degeneration of the retina –pigmentary retinopathy. In addition, patients can have cardiac conduction defects, elevated cerebrospinal fluid (CSF) protein and ataxia. Other neurological abnormalities such as deafness, dementia and endocrine abnormalities including growth retardation, short stature, hypothyroidism, hypoparathyroidism and diabetes mellitus, kidney dysfunction and muscle weakness may also be present.6 Complete heart block is known to be the major cause of death in patients with KSS. Our patient had classic manifestations of the syndrome.
Muscle biopsy or mitochondrial genome sequencing confirms the genotypic diagnosis, but is of no significance for treatment and outcome.5,6,7 Approximately 90% of individuals with KSS have a large-scale (i.e., 1.1- to 10-kb) mtDNA deletion that is usually present in all tissues.8 Muscle biopsy from ocular muscles usually show presence of ragged red fibres.9 Treatment is mainly palliative and supportive for the clinical condition | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Kearns T. Retinitis Pigmentosa, External Ophthalmoplegia, and Complete Heart Block. AMA Archives of Ophthalmology. 1958;60:280. [CrossRef] [PubMed]
- Myasthenia Gravis Fact Sheet. National Institute of Neurological Disorders and Stroke. Available from URL: https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Myasthenia-Gravis-Fact-Sheet. Accessed on 13th March 2018.
- Suzuki S, Utsugisawa K, Yoshikawa H. Autoimmune targets of heart and skeletal muscles in myasthenia gravis. Arch Neurol, 2009;6:1334-1338. [CrossRef] [PubMed]
- Nagahashi S, Carvalho S, Fonseca L, Carvalho M, Reed C, Scaff M. Kearns-Sayre “Plus”. Arq Neuropsichiatry1999;57:1017-1023.
- Nasseh E, Tengan H, Kiyomoto H, Gabbai AA. Doençasmitocondriais. Rev Neurociências 2001;9:60-69.
- Limongelli G, Tome-Esteban M, Dejthevaporn C, Rahman S, Hanna M, Elliott P. Prevalence and natural history of heart disease in adults with primary mitochondrial respiratory chain disease. Eur J Heart Fail. 2010;12:114-21. [CrossRef] [PubMed]
- Ramírez-Miranda A, Navas-Pérez A, Gurria-Quintana L, Vargas-Ortega J, Murillo-Correa C, Zenteno J. Detección de deleciones en DNA mitocondrial heteroplásmico por medio de PCR en el Síndrome de Kearns-Sayre. Archivos de la Sociedad Española de Oftalmología. 2008;83:155-160. [CrossRef]
- DiMauro S, Hirano M. Mitochondrial DNA Deletion Syndromes. 2003 Dec 17 [Updated 2011 May 3]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1203/. Accessed on 13th March 2018
- Richard M, Alfredo A. Ocular Myopathies. In: Textbook of Ophthalmology. Available at URL: https://medtextfree.wordpress.com/2011/04/05/chapter-201-ocular-myopathies/. Accessed on 13th March 2018.
DOI: https://doi.org/10.7199/ped.oncall.2019.26
|
| Cite this article as: | | Chaitanya K, Addanki A, Deshpande N, Badani R. Kearns-Sayre Syndrome Misdiagnosed as Myasthenia Gravis. Pediatr Oncall J. 2019;16: 51-52. doi: 10.7199/ped.oncall.2019.26 |
|