Carolina Amaro Gonçalves1, Ana Dias Curado1, Catarina Salgado2, Isabel Esteves3, Filipa Oliveira Ramos4, Anabela Ferrão2.
1Pediatric Department, Centro Hospitalar e Universitário Lisboa Norte - Hospital de Santa Maria, Lisbon, Portugal,
2Pediatric Haematology Unit, Pediatric Department, Centro Hospitalar e Universitário Lisboa Norte - Hospital de Santa Maria, Lisbon, Portugal,
3Pediatric Infectious Disease and Immunodeficiency Unit , Department of Pediatrics, Centro Hospitalar e Universitário Lisboa Norte - Hospital de Santa Maria, Lisbon, Portugal,
4Pediatric Rheumatology Unit, Centro Hospitalar e Universitário Lisboa Norte - Hospital de Santa Maria, Lisbon, Portugal.
ADDRESS FOR CORRESPONDENCE
Carolina Amaro Gonçalves, Avenida Professor Egas Moniz, Hospital de Santa Maria, 1649-028, Lisboa, Portugal
Email: agoncalves7@campus.ul.pt | | Abstract | | Childhood Sarcoidosis is a chronic multisystem pinflammatory disease of unknown etiology. We report a case of a 9-year-old female who presented with nasal bleeding. She started with fever that persisted for 24 days associated with progressive splenomegaly, pancytopenia and generalized lymphadenopathy. An extensive investigation was performed excluding infectious, neoplastic, autoimmune and immunodeficiency causes. Histological examination of a lymph node revealed multiple non-caseous granulomas, which together with the elevation of ACE and multiorgan involvement, suggested the diagnosis of sarcoidosis. She started oral prednisolone and after 4-month of follow-up there was a significant clinical and analytic improvement. This case represents a challenging diagnosis of an uncommon pediatric disease that should be considered in the differential diagnosis of a multisystemic disease with constitutional symptoms even in the absence of pulmonary involvement. | | | | Keywords | | Sarcoidosis, prolonged fever, pancytopenia, splenomegaly, lymphadenopathy | | | | Introduction | Childhood sarcoidosis is a rare chronic multisystem granulomatous disease of unknown etiology characterized by non-caseating granulomas. The clinical spectrum varies according to the organs involved and depends on the age of affected patients.1 Three distinct forms of the disease have been recognized in children: the familial 0form, with autosomal dominant inheritance (Blau syndrome), the early onset form (<5 years) and juvenile sarcoidosis, similar to adult sarcoidosis/classic form. The triad of skin rash, arthritis and uveitis, usually without pulmonary involvement, is more common in early onset sarcoidosis.2 The presentation of the juvenile form, more frequently during adolescence, is commonly characterized by pulmonary parenchymal infiltration and extrapulmonary disease such as lymphadenopathy, cutaneous and ophthalmic disease. Less often, it may involve the liver, spleen, bone marrow, central nervous system (CNS) and kidney.3,4,5
The incidence and prevalence of pediatric sarcoidosis remain unclear due to the high frequency of subclinical and asymptomatic cases.6,7 A Danish study has reported an annual incidence of 2-3 cases per million of childhood population, with lower incidence in children younger than 4 years of age (0.06/100000) and higher (1.02/100000) in children aged 14-15 years.8
We describe a case of a prolonged fever of unknown origin, progressive splenomegaly and pancytopenia, illustrating the importance of considering this diagnosis even when there is no pulmonary involvement.
| | | | Case Report | We report a case of a 9-year-old female, second child of non-consanguineous parents from Brazil, born and residing in Portugal, admitted to the emergency room (ER) with recurrent and persistent epistaxis associated with cough and rhinorrhea. Fever, loss of appetite, fatigue or weight loss were denied. She had no history of abdominal pain, visual complaints, arthritis, cutaneous manifestations or other bleeding manifestations. There was no known epidemiological context.
She had a history of bronchopulmonary dysplasia due to 26 weeks prematurity and recurrent wheezing treated with inhaled fluticasone. She had been followed in pediatric hematology (between 4 and 7 years-old) due to recurrent epistaxis. Coagulation studies showed an increased activated partial prothrombin time (aPTT) caused by factor XII deficiency but without hemorrhagic disease. She is fully immunized according to national immunization program, including BCG vaccine and had no other relevant past medical history. Concerning the family history, she has a 21-year-old cousin with systemic lupus erythematosus and a history of sudden cardiac arrest in a 23 and 25-years old cousins.
Beyond unilateral nasal bleeding treated with cautery, physical examination revealed a painless splenomegaly (2 cm below the ribs) without additional clinical signs. Initial laboratory tests showed microcytic anemia (hemoglobin 9.7 g/dl, mean corpuscular volume 73 ft), thrombocytopenia (14 000/uL), altered coagulations parameters (aPTT 44.5/29s; prothrombin time of 14.8/11.6s) and elevated alanine aminotransferase (AST) of 144 UI/L and aspartate aminotransferase (ALT) of 238 UI/L with normal bilirubin (Table 1). Chest X-ray was normal.
Table 1. Patient Laboratory data on admission, during hospitalization and follow up.
| |
Admission |
D7-8 |
D16-D17 |
D25-D26 |
Hospital discharge D37 |
2 weeks after discharge |
4 moths of follow-up |
| Hematology |
|
|
|
|
|
|
|
| Hb (g/dL) |
9.7 |
8.0 |
6.4 |
8.2 |
8.4 |
9.2 |
11.5 |
| MCV (fL) |
73 |
73.5 |
72 |
73.9 |
72 |
71 |
|
| WBC (µL) |
4600 |
3800 |
2600 |
3900 |
3700 |
4.20 |
7700 |
| Neutrophyls |
2370 |
1440 |
920 |
1460 |
1430 |
1630 |
5430 |
| Lymphocytes |
1500 |
1790 |
1130 |
1530 |
1510 |
1610 |
1710 |
| Monocytes |
660 |
420 |
500 |
830 |
700 |
840 |
520 |
| Platelet (x109/L) |
14000 |
58000 |
98000 |
108000 |
95000 |
106000 |
126000 |
| Coagulation |
|
|
|
|
|
|
|
| PT (s) |
14.8/11.9 |
13.1/11.6 |
|
|
13.6/11.6 |
12.6/11.6 |
|
| APTT (s) |
44.5/29 |
43.6/29.0 |
|
|
37.4/29 |
39.5/29 |
|
| Fibrinogen (mg/dL) |
253 |
306 |
|
|
261 |
|
|
| Biochemistry |
|
|
|
|
|
|
|
| AST (U/L) |
144 |
132 |
77 |
120 |
241 |
632 |
25 |
| ALT (U/L) |
238 |
167 |
88 |
145 |
309 |
982 |
31 |
| GGT (U/L) |
|
69 |
48 |
46 |
53 |
321 |
29 |
| ALP (U/L) |
|
190 |
128 |
|
172 |
263 |
137 |
| LDH (U/L) |
296 |
274 |
251 |
291 |
379 |
522 |
|
| T Bil (mg/dL) |
0.58 |
0.30 |
0.28 |
0.28 |
0.18 |
0.26 |
|
| Ca (mmol/L) |
8.9 |
|
8.8 |
9.1 |
8.9 |
|
10 |
| Urinary Ca (mg/dL) |
|
|
|
|
4.4 |
|
|
| CRP (mg/dL) |
2.14 |
5.08 |
2.96 |
4.11 |
2.52 |
1.58 |
|
| Ferritin (mg/dL) |
36.5 |
32.4 |
23.0 |
|
32.5 |
29.5 |
|
| ESR (mm) |
|
61 |
|
38 |
|
34 |
8 |
| Albumin (g/dL) |
4.4 |
|
3.5 |
|
4.1 |
4.3 |
|
| P.Electrophoresis |
Normal |
|
Normal |
|
|
|
|
| IgG / IgM / IgA (mg/dL) |
868/149/16 |
|
|
|
|
1351/475/13 |
561/210/17 |
| B12 Vitamin (pg/mL) |
|
1189 |
|
|
905 |
|
|
| ACE (U/L) |
|
|
182 |
188 |
|
220 |
61 |
ACE: angiotensin-converting enzyme, ALP: alkaline phosphatase, Ca: calcium, CRP: C-reactive protein, ESR: erythrocyte sedimentation rate, Hb: hemoglobin, LDH: lactate dehydrogenase, MCV: mean corpuscular volume, PCT: procalcitonin, PT: prothrombine time, T Bil: total bilirubin, WBC: white blood cell.
Due to severe thrombocytopenia, active severe nasal bleeding and good general appearance, the diagnosis of immune thrombocytopenia was considered and she was treated with intravenous immunoglobulin (IVIG) 800 mg/kg. Anemia was assumed in the context of recurrent and severe bleeding. She was admitted to the Pediatric Hematology Unit in a tertiary pediatric center for further investigation.
On the second day of hospitalization, she started fever (max 39.5ºC) with a persistent twice daily pattern over 24 consecutive days. Inflammatory markers were increased (maximum parameters: C-reactive protein 5.06 mg/dL, erythrocyte sedimentation rate 62 mm) but ferritin levels were always in the normal range (Table 1).
Progressive splenomegaly was verified during the hospitalization (max 8 cm palpable below the ribs). The abdominal ultrasound confirmed a massive splenomegaly of 16.5 cm with mild hepatomegaly and celiac trunk, splenic hilum and lumbar aortic lymphadenopathy. Liver function tests reached a maximum of AST 632 U/L, ALT 982 U/L, gamma-glutamyltransferase (GGT) 321 UI/L, with normal bilirubin and alkaline phosphatase.
Due to development of pancytopenia (minimum Hb 6.4 g/dL, leucocytes 2 600/µL, neutrophils 920/µL, lymphocytes 1130/µL, platelets 14 000/µL with normal monocyte, eosinophil and basophil count), needing erythrocyte transfusion, we performed a bone marrow aspirate and biopsy. It revealed a hypercellular bone marrow with normal morphology and maturation and no abnormal cells. Because of a dubious microscope visualization of amastigote forms of leishmania, she was treated with liposomal amphotericin B and complete the entire treatment until serology and nucleic acid amplification test for Leishmania donovani were negative.
Despite meeting criteria for hemophagocytic syndrome (persistent fever; pancytopenia; splenomegaly; hypertriglyceridemia, max 319 mg/dL; increased soluble CD25 receptor, 10 300 pg/mL – reference range: 458-1997 pg/mL), it was decided not to start treatment because she had persistently normal ferritin (max 46 mg/dL) (see Table 1) with preserved general status.
The etiological investigation excluded the main infectious causes: negative serology for Human immunodeficiency virus 1 and 2, Hepatitis A, B and C virus, Cytomegalovirus (CMV), Epstein-Barr virus (EBV), Parvovirus, Herpes-simplex virus 1 and 2, Toxoplasmosis, Brucellosis, Leptospirosis, Bartonella, Coxiella burnetti and Leishmaniasis. EBV and CMV viral load tests in peripheral blood were negative. Nucleic acid amplification test for Leishmania donovani in medullary blood, Plasmodium in blood smear and Interferon-Gamma Release Assay (IGRA) were also negative. She had a positive serology for SARS-CoV-2 compatible with a known asymptomatic SARS-CoV-2 infection 3 months before. Multiple samples of aerobic and anaerobic blood cultures were all negative.
Primary immunologic work-up included immunoglobulin electrophoresis that revealed a mild deficit of 15 mg/dL IgA, with mild hyper IgG (1309 mg/dL) and hyper IgM 433 mg/dL, although it was measured 1 month after post-IVIG infusion. IgG4 was normal.
Beside a positive lupus anticoagulant, the remain autoimmune study that included anti-cardiolipin, anti-beta2-glycoprotein, anti-nuclear antibodies (ANAs), double-stranded DNA (dsDNA), complement, Coombs’s test, antineutrophil cytoplasmic antibodies (c-ANCA and p-ANCA), antimitochondrial antibodies (AMA), anti-smooth-muscle antibody (ASMA) and anti-liver-kidney antibody (LKM) were negative.
Echocardiogram excluded endocarditis and electrocardiography was also normal. The ophthalmologic evaluation did not reveal ocular manifestations of disease.
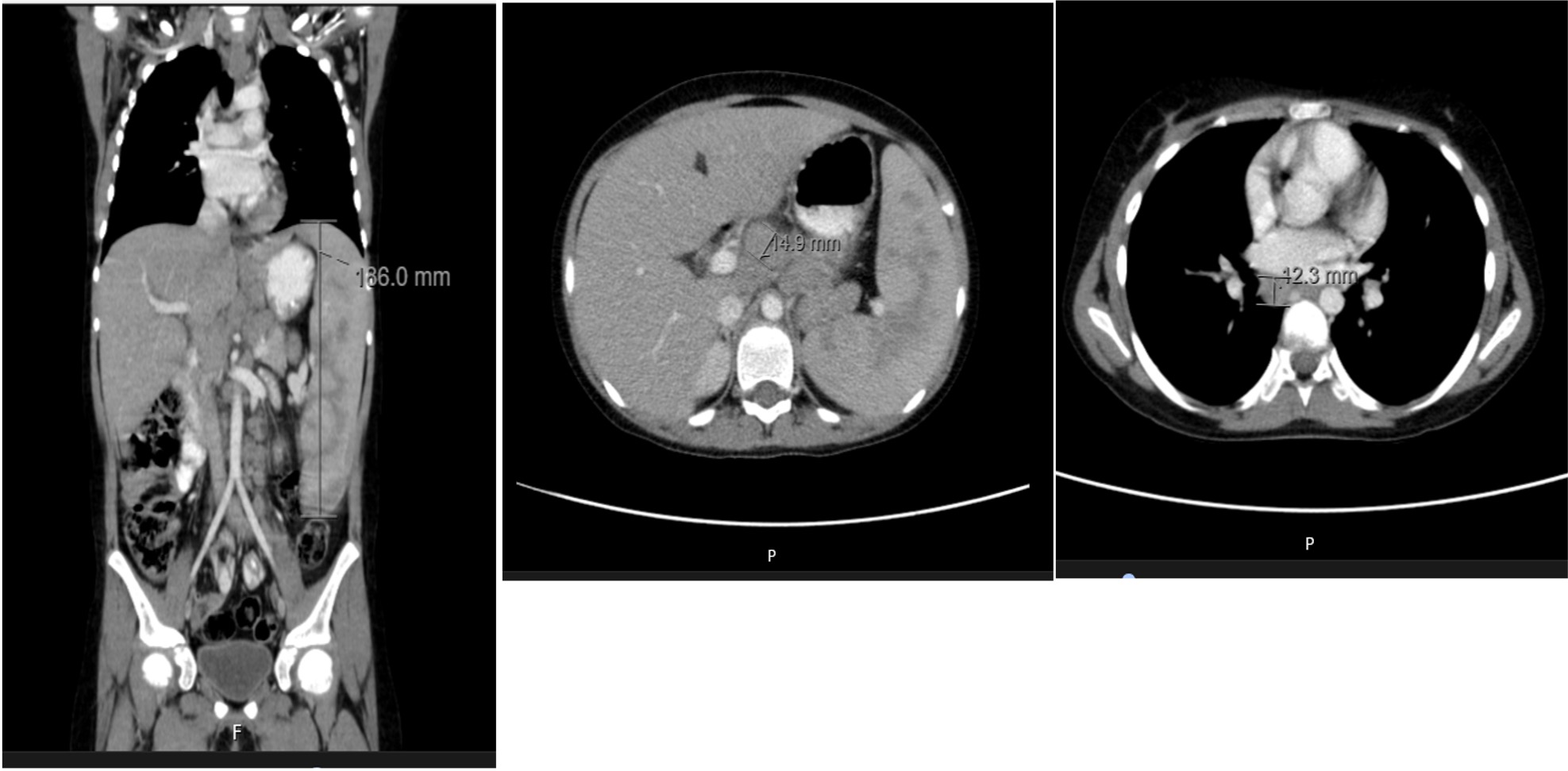
A thoracic-abdominal-pelvic computed tomography (CT) confirmed a mild hepatomegaly and a massive splenomegaly (18.6 cm) and revealed generalized lymphadenopathy with axillary and bilateral supraclavicular lymphadenopathies, right infracarinal, right tracheobronchial and multiple abdominal adenopathies in the celiac trunk territory, splenic hilum, hepatic and lumbo-aortic hilum and in the primitive iliac chains. CT did not reveal involvement of lung parenchyma. (Figure 1).
Figure 1. CT scan without contrast. A - marked heterogeneous splenomegaly (186 mm) and mild homogeneous hepatomegaly. B - multiple abdominal adenopathies with a 15 mm lymph node in celiac trunk. C - mediastinal lymphadenopathy with a 12.3 mm and tracheobronchial adenopathy.
Autoimmune lymphoproliferative syndrome (ALPS) was considered as a possible diagnosis due to clinical course and a high B12 vitamin level. Immunophenotyping was performed by flow cytometry in peripheral blood which revealed 4.2% T cells with αβ receptor and absence of CD4 and CD8 expression in total circulating lymphocytes and a slight reduction of CD4 and CD8+ T subpopulations with naive phenotype and marked expansion of activated cells, as well as terminally differentiated CD8+ T cells. Although this result was compatible with ALPS, a genetic panel by whole exome sequencing excluded mutations in the following genes: ADA2, AICDA, CASP10, CASP8, CD40LG, CTLA4, FADD, FAZ, FASLG, ITK, KRAS, LRBA, MAGT1, NRAS, PRKCD, RASGRP1, SH2D1A, STAT3, WAS.
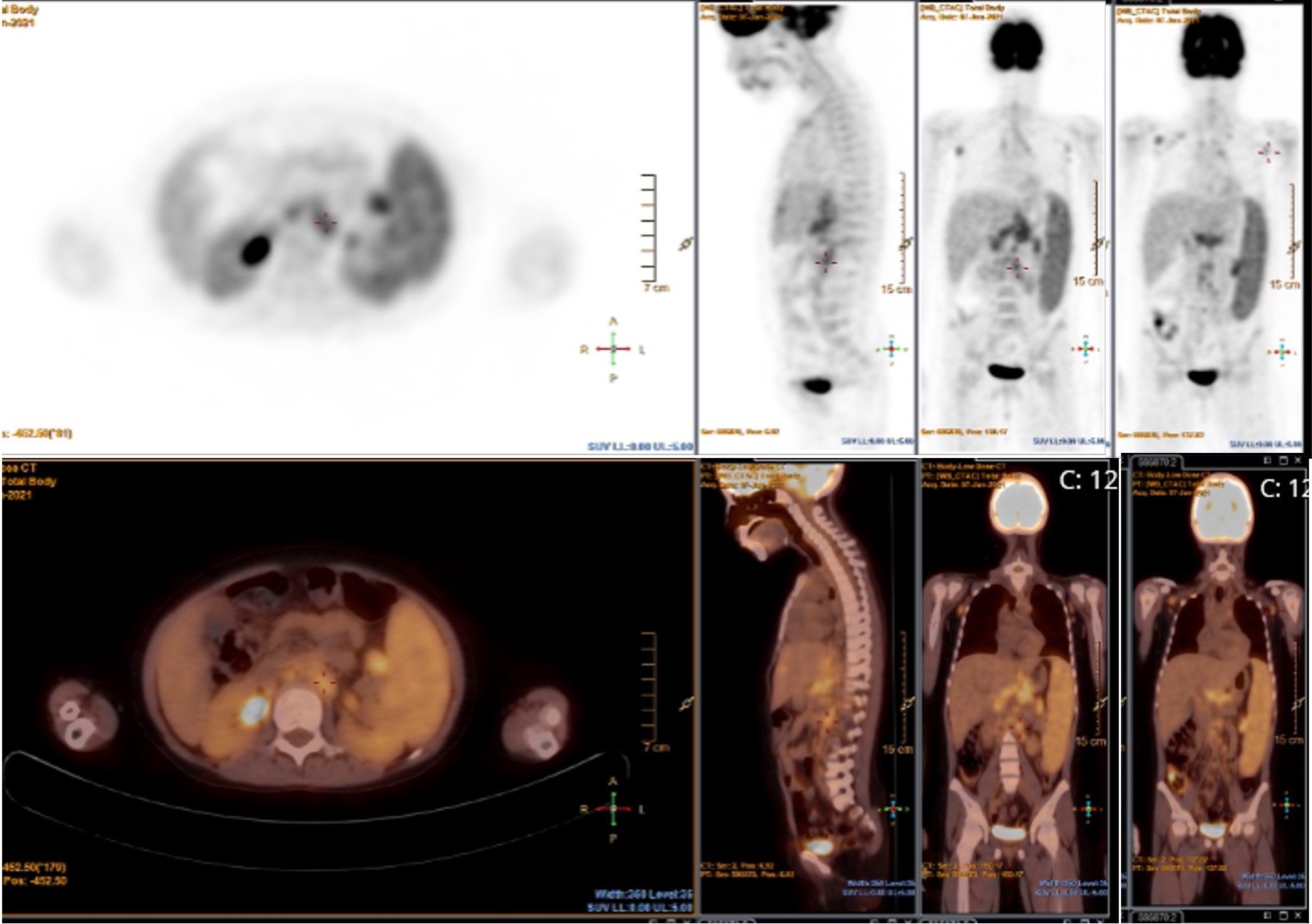
We performed a Positron Emission Tomography (PET) study that evidenced multiple adenopathies with low to moderate metabolic expression in supra and infradiaphragmatic topography (bilateral axillary, mediastinal, right internal mammary chain, paraesophageal, celiac and splenic hilar, lumboaortic and bilateral inguinal) associated with massive homogeneous splenomegaly with higher metabolism than the liver (Figure 2).
Figure 2. PET 18F-FDG showed multiple adenopathies with mild to moderate metabolic expression associated with massive homogeneous splenomegaly.
To rule out a lymphoproliferative disease, an excisional lymph node biopsy was performed. The anatomopathological result revealed numerous epithelioid granulomas sarcoid-like without necrosis. Microorganism investigation by histochemistry (Periodic acid-Schiff stain, Giemsa and Ziehl-Neelsen) was negative (Figure 3). Lymph node immunophenotyping was normal.
We verified an increased plasma concentration of ACE (182-220 U/L). Serum and urinary calcium were normal.
Figure 3. A- Lymph node biopsy with conserved general architecture (H&E, 20X) with follicular hyperplasia (?) and presence of numerous paracortical epithelioid non-caseating granulomas (*). B- Magnification 40X, H&E. C- Epithelioid granuloma with a syncytial arrangement, multinucleate cells with a vast and eosinophilic cytoplasm and vesicular nuclei without necrosis – sarcoid-type granuloma (H&E, 400X).

Given the likely diagnosis of sarcoidosis, we performed further pulmonary tests: pulmonary function tests were normal and analysis of broncho-alveolar lavage (BAL) showed 70% macrophages, 4% neutrophils and 26% lymphocytes (CD4/CD8 ratio was not performed due to insufficient bal fluid quantity).
According to diagnosis of sarcoidosis, corticosteroid therapy with oral prednisolone 1 mg/kg/day (30 mg/day) was started, with a regular clinical and analytical outpatient monitoring.
After 4-month of follow-up, there was a significant clinical and analytic improvement that allowed a gradually reduction of prednisolone after 4 weeks of therapy. There was regression of hepatomegaly, splenomegaly decreased to only 2cm below the ribs and there was no recurrence of the fever. Anemia and leucopenia resolved remaining only a mild thrombocytopenia. Liver enzymes returned to normal values and ACE is progressively decreasing.
At this moment, she is clinically stable, without signs or symptoms of disease recurrence, maintaining a low-dose prednisolone (0.25 mg/kg/day).
| | | | Discussion | Sarcoidosis is a rare chronic multisystemic granulomatous disease, poorly described in pediatric population. It is a challenging diagnosis due to the absence of specific criteria or markers of the disease as well as the variable clinical spectrum from asymptomatic to severe multiorgan disease.9
Clinical manifestations can be variable and nonspecific since it can affect any organ or system. Although pulmonary involvement is observed in most of the cases in juvenile and adult forms, the association of constitutional symptoms and extrapulmonary manifestations without pulmonary manifestations is infrequent. Isolated presentation of extrapulmonary disease alone is also rare.4,5
In our patient the first presenting symptom was severe nasal bleeding probably resulting from severe thrombocytopenia which motivated ER admission. All other symptoms, such as fever, progressive splenomegaly and pancytopenia, appeared progressively throughout the hospitalization allowing an earlier diagnostic investigation to reach the diagnosis.
Constitutional symptoms are often attributed to infectious, immune-mediated or lymphoproliferative diseases. Fever is frequent at the presentation of sarcoidosis in pediatric age opposing to adults.8
Bone marrow aspirate and biopsy was performed to exclude neoplastic, infiltrative or infectious diseases with medullary involvement. The exam was normal with no abnormal hematopoietic cells, no suggestive images of granulomas, nor safe evidence of microorganisms. Given the initial doubtful observation of amastigotes in the bone marrow and compatible clinical symptoms of visceral leishmaniasis, in the absence of an alternative diagnosis, it was decided to treat empirically with Amphotericin B without clinical improvement. Serological and nucleic acid amplification tests for Leishmania Donovani were both negative ruling out this diagnosis.
Most frequent organs affected in Sarcoidosis are the lungs, lymph nodes, skin, eyes and often liver, spleen. Less frequently it can involve musculoskeletal system, kidney, brain, heart and deregulation of calcium metabolism.5,9,10
Cytopenias in sarcoidosis, when present, may reflect the toxicity due to bone marrow infiltration by granulomas, as well as indirectly from the variety of cytokines release.4 Documented bone marrow involvement in adults is rare10,11 and in children there are only a few case reports.13,14 Estimated incidence of granulomas in bone marrow biopsies is low. Brackers et al. showed that the overall incidence of bone marrow epithelioid granuloma was only 0.59% and sarcoidosis accounts for up to 21% of these cases.15 Anemia and leukopenia are the clinical presentations in 4%-40% of the sarcoidosis cases.4 In this case, pancytopenia might be explained by both hypersplenism and medullary involvement, even in the absence of bone marrow granulomas.
Hepatosplenic involvement in sarcoidosis can occur in up to 43% of patients in the course of the disease.3,5,16 However, clinical manifestations of this involvement are uncommon and massive splenomegaly as an initial manifestation is rare.17,18,19 In hepatic sarcoidosis, liver transaminases levels are usually normal or moderately increased (2-3 times), 50-70% have increased GGT and/or ALP and, rarely, increased bilirubin.10,20,21 In this case and as reported by Nawfal et al.21, transaminase values were more than 10 folds the normal range with an increased GGT but normal ALP and total bilirubin. Other causes of hepatic cytolysis, namely infectious, autoimmune and toxic were excluded.
CT showed a generalized lymphadenopathy, with involvement of mediastinic and abdominal territories, metabolically active in the PET scan, leading to consideration of the following diagnoses: lymphoma, tuberculosis, ALPS or sarcoidosis.
Lymphatic system involvement is one of the most common manifestations in pediatric sarcoidosis and may reach 40-70% of cases, especially in older children.3,7,10 Peripheral adenopathies are generally discrete and non-painful. Although our patient had generalized lymphadenopathy, they were not clinically evident. Mediastinal and mesenteric territory, especially celiac trunk, are characteristic locations.22
As the hypothesis of ALPS was considered, we performed immunophenotyping that identified alpha beta double-negative T cells. An increased B12 vitamin levels were also compatible with ALPS, but genetic study excluded the most probable genetic causes for this diagnosis.
It was decided to perform an excision of a peripheral and easily accessible lymph node. We did not perform any other more invasive procedures, such as liver or splenic biopsy, considering the risk of hemorrhage. Lymph node immunophenotyping was normal and histological examination of the ganglion revealed multiple non-caseous granulomas, which together with the elevation of ACE, multiorgan involvement and after excluding infectious, neoplastic, autoimmune and immunodeficiency causes, suggested the diagnosis of sarcoidosis.
There are no specific tests available for sarcoidosis and the diagnosis currently requires the histological demonstration of non-caseous granuloma in one or more organs and consistent clinical and imaging findings, after a rigorous investigation to exclude alternative diagnosis, especially indispensable in children in whom pediatric sarcoidosis remains a diagnosis of exclusion.23,24,25,26
Typical noncaseating epithelioid granuloma consists of clearly circumscribed epithelioid histiocytes
and multinucleate giant cells with lymphocyte border.4,23 Elevated levels of ACE are found in more than 50% of children with late-onset disease although this is not specific nor sensitive.4,26,27
Other granulomatous diseases share histological characteristics. Infectious diseases are the most common. Alternative diagnosis includes tuberculosis and other mycobacteriosis, toxoplasmosis, leishmaniasis, brucellosis and infections by other bacteria and fungus. Non-infectious causes are less frequent and include vasculitis, neoplasia, systemic immune diseases, immunodeficiency, drug/chemical-induced granulomatous reactions.28 In this patient, after we excluded other causes of systemic disease, the diagnosis of sarcoidosis with apparently exclusive extrapulmonary expression became the most likely.
Although chest X-ray and chest CT did not demonstrate involvement of the pulmonary parenchyma, the involvement of mediastinal lymphadenopathy was evident. She performed pulmonary function tests that were normal.
Pulmonary involvement is variable. Symptoms are usually mild including dry cough, dyspnea or thoracalgia. However, most patients are asymptomatic.29 Restrictive lung disease is the typical alteration found in pulmonary function tests. Chest X-ray is frequently normal, but high-resolution CT reveals abnormalities in 95% of children, including nodules with frosted glass opacities to hilo-mediastinical lymphadenopathies.3,4,6,7,29,30
Bronchofibroscopy revealed the presence of a predominance of macrophages. It was not possible to evaluate the CD4/CD8 ratio of bal which may be a useful tool in the diagnosis of sarcoidosis, that although highly specific are not sensitive.31
Pediatric sarcoidosis long-term studies of sarcoidosis in pediatric age are lacking due to its rareness. Therefore, the prognosis remains uncertain with high variability in disease extent and severity. There is no curative therapy and treatment should be individualized because a large percentage of cases have a self-limited course.
Indications for absolute therapy include progressive or symptomatic lung disease or changes in pulmonary function including restrictive pattern; ocular involvement; disfiguring skin disease; cardiac sarcoidosis; sarcoidosis with CNS involvement, significant hypercalcemia and symptomatic hepatosplenic involvement.1,23,24
Treatment is not standardized for pediatric age but, like adults, corticosteroids are the first-line treatment at a dose of 1-2 mg/kg/day for 4-8 weeks as induction therapy.5,6 Second-line drugs include methotrexate, TNF-α antagonists, cyclophosphamide, mycophenolate mofetil and tacrolimus.6,7,10,13,29
In our patient it was decided to start therapy because of the maintenance of cytopenias with massive splenomegaly and hepatic involvement with progressive elevation of transaminases. After treatment, she had a favorable clinical course with total regression of splenomegaly and normalizations of liver enzymes with a residual mild thrombocytopenia. Both the serum levels of ACE and ESR decreased, which can be helpful tools, although non-specific, in monitoring the disease activity.27
In conclusion, pediatric sarcoidosis is a challenging diagnosis due to the lack of specific signs and symptoms that mimics other diseases.32 Although rare in children, sarcoidosis should be considered in the differential diagnosis in the case of diseases with multiorgan involvement such as lymphadenopathy, hepatosplenomegaly and cytopenia associated with systemic symptoms such as fever, even in the absence of pulmonary involvement and after the exclusion of other diseases. | | | | Financial Disclosure | | The authors declared that this study has received no financial support. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest | | The authors have no conflict of interest to declare. | | |
- Costabel U, Hunninghake GW. ATS/ERS/WASOG statement on sarcoidosis. Sarcoidosis Statement Committee. American Thoracic Society. European Respiratory Society. World Association for Sarcoidosis and Other Granulomatous Disorders. Eur Respir J. 1999;14(4):735-737. [CrossRef] [PubMed]
- Chiu B, Chan J, Das S, Alshamma Z, Sergi C. Pediatric Sarcoidosis: A Review with Emphasis on Early Onset and High-Risk Sarcoidosis and Diagnostic Challenges. Diagnostics (Basel). 2019;9(4):160. [CrossRef] [PubMed] [PMC free article]
- Pattish EN, Kendig EL Jr. Sarcoidosis in children. Pediatr Pulmonol. 1996;22(3):195-203. [CrossRef]
- Shetty AK, Gedalia A, Sarcoidosis in Children. Curr Probl Pediatr. 2000;30(5): 149-76. [CrossRef] [PubMed]
- Shetty AK, Gedalia A. Childhood sarcoidosis: A rare but fascinating disorder. Pediatric Rheumatol 2008: 6:16. [CrossRef] [PubMed] [PMC free article]
- Nathan N, Marcelo P, Houdouin V for the RespiRare and the French Sarcoidosis groups, et al. Lung sarcoidosis in children: update on disease expression and management. Thorax 2015; 70:537-542. [CrossRef] [PubMed]
- Gedalia A, Khan TA, Shetty AK, Dimitriades VR, Espinoza LR. Childhood sarcoidosis: Louisiana experience. Clin Rheumatol. 2016;35(7):1879-1884. [CrossRef] [PubMed]
- Hoffmann Al MN, Byg KE. Childhood sarcoidosis in Denmark 1979-1994: incidence, clinical features and laboratory results at presentation in 48 children. Acta Paediatr Int J Paediatr. 2004;93(1):30-6. [CrossRef]
- Fretzayas A, Moustaki M, Vougiouka O. The puzzling clinical spectrum and course of juvenile sarcoidosis. World J Pediatr 2011; 7: 103-10. [CrossRef] [PubMed]
- Fauroux B, Clément A. Paediatric sarcoidosis. Paediatr Respir Rev. 2005;6(2): 128-33. [CrossRef] [PubMed]
- Baughman RP, Teirstein AS, Judson MA, Rossman MD, Yeager H, Bresnitz EA, DePalo L, Hunninghake G, Iannuzzi MC, Johns CJ, McLennan G, Moller DR, Newman LS, Rabin DL, Rose C, Rybicki B, Weinberger SE, Terrin ML, Knatterud GL, Cherniak R, Case Control Etiologic Study of Sarcoidosis (ACCESS) research group: Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care. 2001, 164: 1885-1889. [CrossRef] [PubMed]
- Pena-Garcia JI, Shaikh S, Barakoti B, Papageor- giou C, Lacasse A. Bone marrow involvement in sarcoidosis: An elusive extrapulmonary manifestation. J Community Hosp Intern Med Perspect 2019; 9: 150-4 [CrossRef] [PubMed] [PMC free article]
- Al-Mayouf SM, Al-Sonbul A, Al Jumaah S, Al-Hemidan A. Sarcoidosis: a delayed or missed diagnosis in children. Ann Saudi Med. 2006 May-Jun;26(3):220-3. [CrossRef] [PubMed] [PMC free article]
- Al Meshram RM, Gajimwar VS, Gholap S, Jhanwar M. Bone marrow involvement: atypical presentation of early-onset childhood sarcoidosis. Eur J Rheumatol 2020; 7(4): 190-4. [CrossRef] [PubMed] [PMC free article]
- Brackers de Hugo L, French M, Brousssolle C, Seve P. Granulomatous lesions in bone marrow: Clinicopathologic finding and significance in a study of 48 cases. Eur J Intern Med 2013; 24: 468-73 [CrossRef] [PubMed]
- Gunathilaka PK, Mukherjee A, Jat KR, Lodha R, Kabra SK. Clinical Profile and Outcome of Pediatric Sarcoidosis. Indian Pediatr. 2019;56(1):37-40. [CrossRef] [PubMed]
- Viprey M, Donadieu J, Epaud R, et al. Massive splenomegaly and pancytopenia revealing sarcoidosis in a child. Sarcoidosis Vasc Diffuse Lung Dis. 2013;30(2):149-152. Published 2013 Aug 1.
- Sherief LM, Amer OT, Mokhtar WA, Kamal NM, Ibrahim HM. Pediatric sarcoidosis presenting as huge splenomegaly. Pediatr Int. 2017;59(3):366-367. [CrossRef] [PubMed]
- Cobreros-Pérez Á, Galindo-Zavala R, Carazo-Gallego B, Martín-Pedraz L, Núñez-Cuadros E. Systemic sarcoidosis; when splenomegaly is not what it seems. An Pediatr (Engl Ed). 2021;94(1):48-50. [CrossRef]
- Valla DC, Benhamou JP. Hepatic granulomas and hepatic sarcoidosis. Clin Liver Dis. 2000;4(1):269-x. [CrossRef]
- Nawfal G, Budin C, Bouvier R, Lachaux A. Elevated aminotransaminases as the first manifestation of sarcoidosis. Case Rep Med. 2009; 2009:193785. [CrossRef] [PubMed] [PMC free article]
- Warshauer DM, Lee JK. Imaging manifestations of abdominal sarcoidosis. AJR Am J Roentgenol. 2004;182(1):15-28. [CrossRef] [PubMed]
- Culver DA. Sarcoidosis. Immunol Allergy Clin N Am. 2012;32(4):487-511. [CrossRef] [PubMed]
- Cimaz R, Ansell BM. Sarcoidosis in the pediatric age. Clin Exp Rheumatol. 2002;20(2):231-237.
- Costabel U, Ohshimo S, Guzman J. Diagnosis of sarcoidosis. Curr Opin Pulm Med. 2008;14(5):455-461. [CrossRef] [PubMed]
- Milman N, Hoffmann AL, Byg KE. Sarcoidosis in children. Epidemiology in Danes, clinical features, diagnosis, treatment and prognosis. Acta Paediatr. 1998;87(8):871-878. [CrossRef] [PubMed]
- Beneteau-Burnat B, Baudin B, Morgant G, Baumann FC, Giboudeau J: Serum angiotensin-converting enzyme activity in normal children and in those with sarcoidosis. Clin Chem 1990, 36:344-346. [CrossRef] [PubMed]
- James DG,A clinicopathological classification of granulomatous disorders. Postgraduate Medical Journal 2000;76:457-465. [CrossRef] [PubMed] [PMC free article]
- Baculard A, Blanc N, Boulé M, et al. Pulmonary sarcoidosis in children: a follow-up study. Eur Respir J. 2001;17(4):628-635. [CrossRef] [PubMed]
- Nathan N, Sileo C, Calender A, et al. Paediatric sarcoidosis. Paediatr Respir Rev. 2019; 29:53-59. [CrossRef] [PubMed]
- Shen Y, Pang C, Wu Y, et al. Diagnostic performance of Bronchoalveolar lavage fluid CD4/CD8 ratio for sarcoidosis: a meta-analysis. EBioMedicine. 2016; 8:302-8. [CrossRef] [PubMed] [PMC free article]
- Giovannini M, Luzzati M, Ferrara G, et al. Common symptoms for a rare disease in a girl with sarcoidosis: a case report. Ital J Pediatr. 2018;44(1):74. [CrossRef] [PubMed] [PMC free article]
DOI: https://doi.org/10.7199/ped.oncall.2023.31
|
| Cite this article as: | | Gonçalves C A, Curado A D, Salgado C, Esteves I, Ramos F O, Ferrão A. Prolonged fever, pancytopenia and splenomegaly - is it sarcoidosis?. Pediatr Oncall J. 2023;20: 100-105. doi: 10.7199/ped.oncall.2023.31 |
|