Nilay Hakan1, Mustafa Aydin1, Nurullah Okumus1, Aysegul Zenciroglu1, Semra Cetinkaya2.
1Department of Neonatology, Dr. Sami Ulus Maternity and Children Hospital, Ankara, Turkey,
2Department of Pediatric Endocrinology, Dr. Sami Ulus Maternity and Children Hospital, Ankara, Turkey.
ADDRESS FOR CORRESPONDENCE
Dr. Nilay Hakan, Division of Neonatology, Department of Pediatrics, Erzurum Regional Training & Research Hospital, Neonatal Intensive Care Unite, Erzurum, Turkey.
Email: nhakan@hotmail.com | | Keywords | | Martsolf syndrome, microcephaly, congenital cataract, hypogonadism, newborn infant | | | A twenty days old baby was admitted to hospital for investigating central lens opacities. He was born at 39 weeks gestation by spontaneous delivery to a 30-year-old gravida 3, para 3 mother with a 3030 g birth weight. Prenatal and family history was unremarkable, except his parents were first degree cousins. His actual weight was 3660 g (10-25 percentile), length 52 cm (10-25 percentile), and head circumference 33 cm (<3 percentile). There was bilateral central lens opacities, craniofacial dysmorphism (flat occiput, high arched palate, low nasal bridge, mild micrognathia, and low posterior hairline), and microcephaly (Fig. 1). Genitourinary examination revealed bilateral cryptorchidism and micropenis (a stretched penile length of 1 cm, <10 percentile). On laboratory investigations, basal level of follicle-stimulating hormone (FSH) was <0.3 mUI/ml (0.26-3), luteinizing hormone (LH) <0.2 mUI/ml (0.02-0.3), and testosterone 4.9 ng/dl (<3-10). Serum cholesterol level was normal. Human chorionic gonadotropin (HCG) stimulation test demonstrated normal Leydig cell reserve (testosterone 354 ng/dl). However, hcg stimulation could not descend testes to scrotum. Skeletal survey, abdominal and cranial ultrasonographies were all normal. A secundum type atrioseptal defect was detected on echocardiography. Chromosome analysis showed a normal male karyotype, 46XY. But, molecular study couldn't be done. Cranial magnetic resonance imaging (MRI) showed mild enlargement of the sylvian fissures, sulci of the frontal and temporal lobes, whereas hypophysis MRI was normal. Normal responses were obtained on brainstem auditory and visual evoked potentials.
At 45 days of age, he was operated for bilateral congenital cataracts, and thereafter, he underwent second operation for undescended testes by 1 year of age. With increasing age, severe psychomotor retardation has been evident.
Figure 1: Craniofacial abnormalities showing low nasal bridge, mild micrognathia, microcephaly
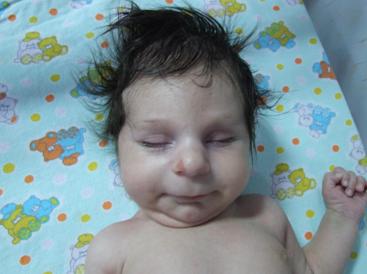
Association of craniofacial dysmorphism, severe psychomotor retardation, cataract, and hypogonadism led to the diagnosis of Martsolf syndrome. It is a rare autosomal recessive syndrome characterized by microcephaly, mental retardation, congenital cataract, hypogonadism, and short stature. In 1978, Martsolf et al (1) first reported two brothers born from consanguineous Jewish parents. In another case report, two brothers, whose parents were unrelated Sephardic Jews, were described to having the findings of Martsolf syndrome. (2) Minor features also seen with this syndrome are brachycephaly, lax finger joints, talipes valgus, a pouting mouth, and maxillary retrusion with slight hirsutism. (1,2) A homozygous missense mutation in RAB3GAP2, the gene for Rab3 GTPase-activating protein, noncatalytic subunit (RAB3GAP2 or KIAA0839), has recently been identified in a consanguineous family with the Martsolf syndrome. (3) Differential diagnosis includes Smith- Lemli- Opitz syndrome (SLOS), oculopalatocerebral syndrome, Lowe's syndrome (LS), and galactosemia. Because the cataract is being rare in SLOS, and serum cholesterol level being normal in our patient, we excluded this syndrome. (4) Absence of a cleft palate, cerebral atrophy and persistent hyperplastic primary vitreous with microphthalmia, which are cardinal features of the oculopalatocerebral syndrome excluded this entity. Lowe's syndrome was excluded by absence of the findings of Fanconi type renal tubular dysfunction. (5) Galactosemia was excluded by showing normal activity of the enzyme of galactose-1-phosphate-uridyltransferase (GALT).
Conflict of Interest and Financial Disclosure Statement: None
Note: This manuscript was presented in the abstract book of 2nd International Congress of Union of European Neonatal and Perinatal Medicine (UENPS). | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Martsolf JT, Hunter AG, Haworth JC. Severe mental retardation, cataracts, short stature, and primary hypogonadism in two brothers. Am J Med Genet 1978; 1:291-9. [CrossRef] [PubMed]
- Sanchez JM, Barreiro C, Freilij H. Two brothers with Martsolf's syndrome. J Med Genet 1985; 22:308-10. [CrossRef] [PubMed]
- Aligianis IA, Morgan NV, Mione M, Johnson CA, Rosser E, Hennekam RC, et al. Mutation in Rab3 GTPase-activating protein (RAB3GAP) noncatalytic subunit in a kindred with Martsolf syndrome. Am J Hum Genet 2006; 78:702-7. [CrossRef] [PubMed]
- Porter FD. Smith-Lemli-Opitz syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet 2008; 16:535-41. [CrossRef] [PubMed]
- Kanik A, Kasap-Demir B, Atesli R, Eliacik K, Yavascan O, Helvaci M. A novel OCRL1 gene mutation in a Turkish child with Lowe syndrome. Turk J Pediatr 2013; 55:82-5. [PubMed]
DOI: https://doi.org/10.7199/ped.oncall.2014.9
|
| Cite this article as: | | Hakan N, Aydin M, Okumus N, Zenciroglu A, Cetinkaya S. MARTSOLF SYNDROME IN A NEWBORN INFANT. Pediatr Oncall J. 2014;11: 22. doi: 10.7199/ped.oncall.2014.9 |
|