Roshni Sonawane.
Department of Pediatrics, Rockingham General Hospital, Western Australia.
ADDRESS FOR CORRESPONDENCE
Dr. Roshni Sonawane, Department of Paediatrics, Rockingham General Hospital, Elanora Drive, Cooloongup, WA 6168, Australia.
Email: doc_roshnis@yahoo.com | | Abstract | | Abetalipoproteinemia (ABL) or Bassen–Kornzweig Syndrome is a rare autosomal recessive disorder of lipid metabolism caused by mutations in the microsomal triglyceride transfer protein gene (MTP). There is resultant impairment in the process of lipid transportation mediated by apoprotein B-containing lipoproteins (Apo-B) in the intestine and liver. The characteristic features are very low plasma levels of these lipoproteins and resultant low levels of, triglyceride (TG), cholesterol and fat-soluble vitamins. The salient clinical features are fat malabsorption and acanthocytosis during infancy and neuro-ocular complications during adolescence. Disease surveillance involves monitoring for ophthalmologic, neurologic, hematologic and hepatic complications. The mainstay of treatment is a low-fat diet and high dose vitamin supplementation. Early diagnosis and management can prevent disease progression. This review is a summary of the genetic, metabolic and clinical features including the available management options. | | | | Keywords | | Abetalipoproteinemia (ABL), Microsomal triglyceride transfer protein gene (MTP), Apolipoprotein-B (Apo-B) | | | | Introduction | Abetalipoproteinemia (ABL) is an inherited metabolic disorder with a heterogeneous clinical presentation. Estimated frequency of the disease is 1 in 10,00,000. (1,2) The name “abetalipoproteinemia” is derived from the typical lack of lipoproteins with beta-electrophoretic mobility on electrophoresis. (3) Bassen and Kornzweig first described the clinical features of abetalipoproteinemia in 1950. (4,5)
Etiology
The causative factor is mutations in the microsomal triglyceride transfer protein gene (MTP) (1), localized to chromosome 4q22-24. (6) About 30 mutations in MTP are known of which point mutations (missense, nonsense and splicing) predominate. (1,4,6) MTP plays a major role in the lipoprotein assembly. Mutation in the MTP gene affects the Apo-B synthesis and leads to its rapid catabolism. This disrupts the intracellular assembly and subsequent secretion of Apo B-containing lipoproteins. The overall outcome is an impairment in the absorption of lipids and lipid-soluble nutrients and vitamins. (7)
Fig: 1 Lipoprotein Assembly and MTP
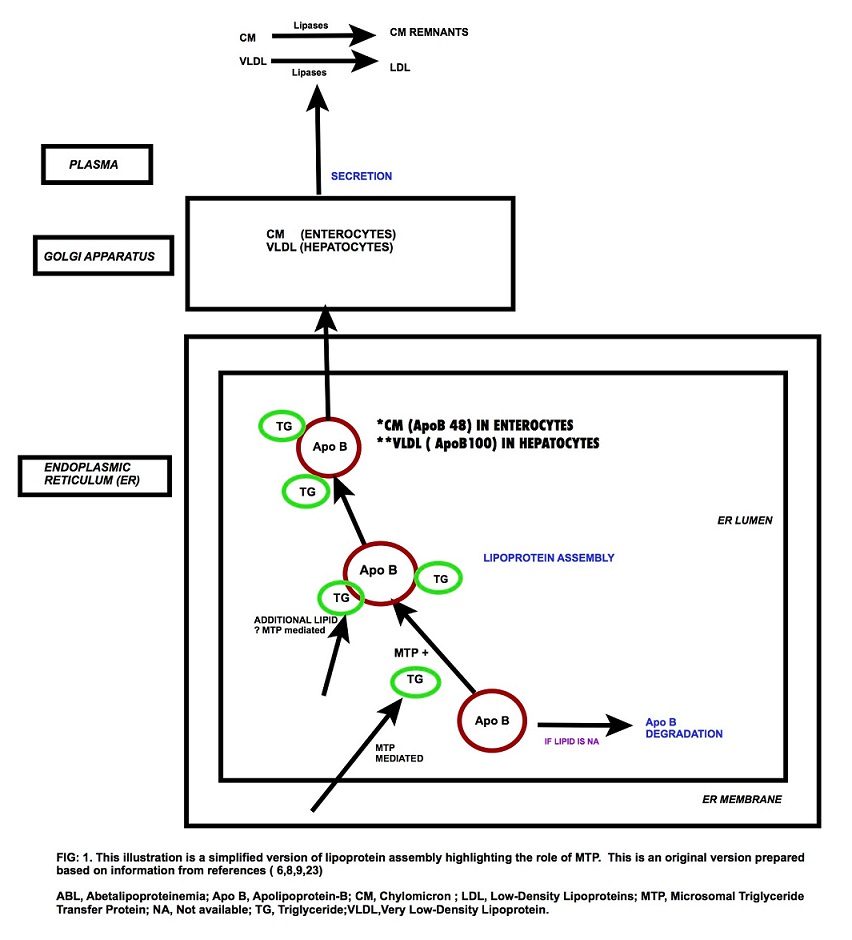
Pathogenesis
In ABL patients, the levels of vitamin E are significantly reduced while vitamins A, D, and K levels do not drop as much. An answer for this can be that vitamin E predominantly relies on Apo-B mediated intestinal absorption, plasma transport, and tissue distribution (Fig 1). (6) Almost all of the vitamin E in enterocytes seems to incorporate into chylomicrons. (4) On the other hand, vitamin D is not dependent on lipoproteins for their absorption or carriage in plasma and, although vitamin A and K initially follow the journey through intestine and liver via the conventional lipoprotein way, subsequently they have their independent transportation systems in the circulation. (8) Vitamin E deficiency is, thus, a chief factor in the pathogenesis of ABL. Albeit, some features of vitamin A and vitamin K deficiency are also present and while vitamin D deficiency is not a consistent feature among ABL patients (6), low serum ionized calcium, vitamin D and bony abnormalities (rickets, osteomalacia) have been described in few cases. (7)
Vitamin E is a free radical scavenger and protects the mitochondrial membranes. In vitamin E deficiency, lipoperoxidation leads to the formation of pigment lipofuscin. Lipofuscin deposits have been noted in multiple organ tissues like skeletal striated muscle, liver, myocardium, spinal cord, and retina in patients with ABL, implying underlying vitamin E deficiency. (3) This is supported by animal studies too. (9) Predominantly, there is axonal degeneration of the spinocerebellar tracts and demyelination of the fasciculus cuneatus and gracilis (6,8,10) including large nerve fibres. (8) In retina, there are reduced number of photoreceptor cells (8,11) Another hypothesis is that the reduced plasma lipid levels change the structural integrity of the membranes in the retina. (11) Insufficient levels of essential fatty acids (EFA) or other nutrients needed to maintain normal retinal function may be other hidden contributing factors (3)
Figure 2: Vitamin E metabolism
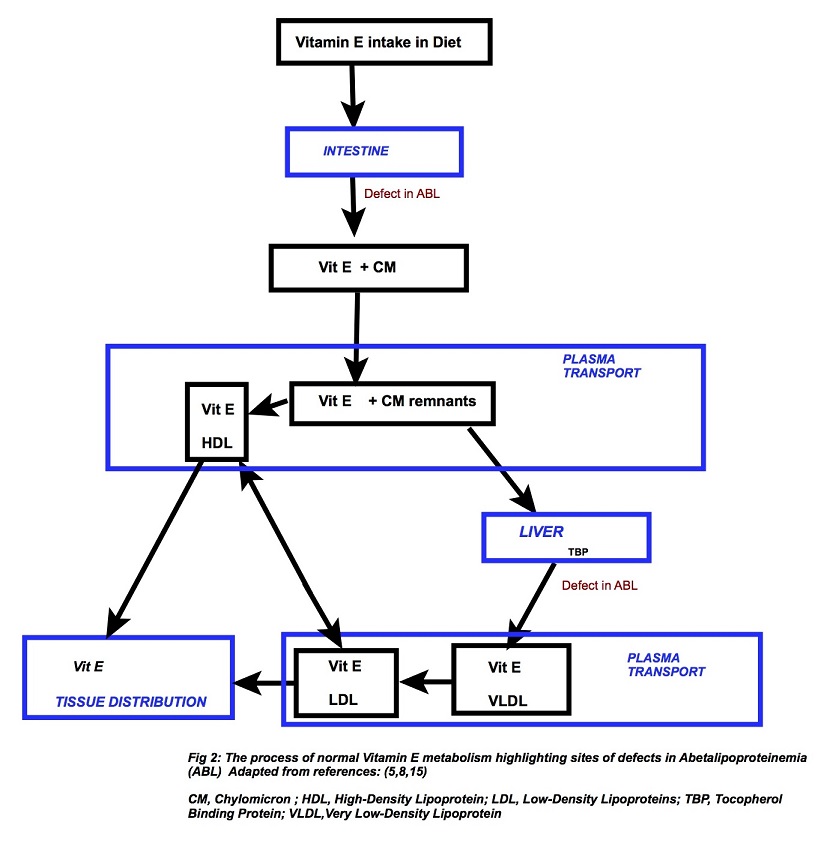
Clinical Presentation
The salient elements of ABL presentation are listed in Table: 1. The clinical picture described in ABL is of a young child with ‘celiac-like' syndrome and spinocerebellar ataxia.
Table:1 Overview of the manifestations in Abetalipoproteinemia (ABL)
| |
PRESENTATION |
| |
History of consanguinity |
| Infancy |
Gastrointestinal: (Pseudoceliac syndrome)*
• Fat malabsorption (Steatorrhoea, FTT) ^
• Vomitting
• Endoscopic examination (on a normal fat diet):
• “gelee blanche” or "white hoar frosting" appearance
• microscopy: lipid laden enterocytes
Hematological:
• Full Blood count: Anaemia*
• Peripheral smear: Acanthocytosis * ^
• ESR: low
• Coagulation abnormalities
Biochemical: (abnormal lipid profile) ^
• Low levels of Apo-B, VLDL, CM, LDL and Cholesterol
• Agarose electrophoresis: Absent beta-lipoprotein band (10)
|
| 1st -2nd decade |
1) Neurological ^ ^
Posterior column Neuropathy:
• Loss of propioception
• Romberg poisitive
• Loss of Deep Tendon Reflexes**
Pyramidal tracts:
• Spastic gait
• Babinski positive
• Severe spasticity by 3rd-4th decades
Cerebellar Pathways:
• Ataxia
• Dysmetria
Myopathy: Generalised muscular weakness leads to
• kyphoscoiosis,
• Lordosis,
• pes cavus
• untreated: progress to immobility
• Complex conduction reduction in the distal motor fibers,
signs of demyelination or distal regeneration
can be of diagnostic value. (26)
2) Ocular: ^ ^
• Ophthalmoplegia
• Ptosis
• Nystagmus
• Strabismus
• Atypical Retinal Dystrophy:
• Nyctalopia***
• Loss of colour vision***
• Defect in visual acuity
• Constricted visual fields
• Gradual expanding scotomas
• Progressive loss of vision
• Corneal ulcers
• Fundoscopy changes (3):
• Atypical retinal pigmentation
• Angiod streaks
• Electroretinogram: markedly reduced (3)
• Fluorescein angiography
• Initial stage: Pigmentary changes in the posterior fundus-small
black spots surrounded by hyperfluorescent halos
• Later stage: salt and pepper appearance and small areas
of chorioretinal atrophy in the perimacular region
• Advanced stage: gradually increasing areas of atrophy. (13)
• SSEPs (Somatosensory Evoked Potentials) and VEPs (Visual Evoked Potentials): altered (14)
|
| Rare presentations reported in ABL patients |
Hepatic:
• Steatosis
• Elevated serum transaminases
• Very rarely: fibrosis and cirrhosis
Cardiac: Sudden cardiac death
Immunological: CD1 deficiency
Malignant potential: Gastrointestinal and Neurological malignancies have occurred
|
* First feature of ABL, **First feature of of CNS presentation in
ABL, ***First feature of Ocular presentation in ABL, ^ recorded in
almost all ABL patients (2,6,9,11), ^ ^ develop insidiously in most ABL
patients (if not all), during the first two decades. (6,11,) (Additional
References used to formulate above table: 4, 6, 13, 14)
Gastrointestinal: The steatorrhea and growth faltering are seen following exposure to a fat-rich diet after birth. There may be associated other gastrointestinal symptoms like vomiting and abdominal distention. These symptoms usually respond to lowering fat intake, which is eventually adopted by the patient because of the ongoing fat intolerance. (7) The associated chronic diarrhea then affects absorption of other nutrients (proteins and carbohydrates). There is ensuing malnutrition and failure to thrive. Endoscopic examination conducted while on fat diet reveals a "gelee blanche" or "white hoar frosting" appearance of the intestinal epithelium. (9) The microscopy of biopsied specimens shows lipid-laden enterocytes. (12) The structural integrity of the villi remains intact. (9) Fat-soluble vitamin deficiencies ensue eventually. (9)
Liver: Liver disease is an unusual feature of ABL. When involved, hepatic manifestations in ABL include steatosis, hepatomegaly, and elevated serum transaminases. Chronic retention of lipids in the hepatocytes after impaired lipid efflux is the supposed process (11). Vitamin A therapy too can iatrogenically exacerbate the liver disease. (6,13) Concerns regarding medium-chain triglyceride (MCT) supplementation causing worsening of hepatic steatosis to progressive fibrosis have been raised. The postulated method is stimulation of endogenous triglycéride synthesis. (11)
Ophthalmology: Ocular manifestations like ophthalmoplegia, ptosis, nystagmus, and anisocoria can be present. (3,13,) The salient feature is an atypical pigmentary retinopathy resembling retinitis pigmentosa (13) It initially presents with nyctalopia and loss of colour vision in most cases. (3) Decline in visual acuity and visual fields develop subsequently. (3) Gradually expanding annular scotomas with macular sparing follow. There is a progressive reduction in the vision (14) and failing intervention can ultimately lead to blindness. (4) Fundoscopy, electroretinogram, and fluorescein angiography can aid to detect the characteristic changes (Table:1). These changes have been viewed in few asymptomatic ABL patients (3,9) as well.
Central Nervous system: As discussed above, the neurological lesion involves posterior columns, the pyramidal tracts, and cerebellum (4,9,14,15) resembling Friedrich’s ataxia. It is characterized by loss of deep tendon reflexes (as the first neurological sign), unsteady gait, generalized muscular weakness (leading to kypho-scoliosis, lordosis, and pescavus), (14) a positive Babinski sign and loss of proprioception. If left untreated, there is worsening of the neurological features leading to severe impairment in mobility requiring assistance, during the 1st and 2nd decade of life. (14) Although few ABL patients have had developmental delays or intellectual disabilities, intelligence is usually unaffected in ABL. (16)
Hematological: Acanthocytosis (crenated RBCs seen on peripheral smear) and anemia may be the first signs of this disorder. (17) Low levels of cholesterol in the erythrocyte membrane (17) lead to the structural disturbances and inhibits rouleaux formation. (14) The erythrocyte sedimentation rate (ESR) is thus lowered. Acanthocytes form more than half of the circulating erythrocytes in ABL patients. (6,12) The peripheral smear should be prepared fresh, preferably avoiding anticoagulants like EDTA. Delay in processing over 6 hours with EDTA allows the formation of regular uniform spicules on the surface of almost all the RBCs and the lymphocytes develop lobulated nuclei. This can be a distinguishing feature, as in ABL many normal red cells are also present. (18) The projected etiology of anemia is hemolysis following vitamin E deficiency. Other causes can be, secondary nutrient deficiencies (iron or folate). Coagulation abnormalities are due to deficiency of vitamin K- dependent coagulation factors (9) showing increased prothrombin time (PT) and increased international normalized ratio (INR). Few patients with ABL have had gastrointestinal bleeding. (6)
Immunology: Zeissig et al discussed the role of MTP in regulating the CD1 family of lipid antigen-presenting molecules. (19) Correspondingly, they highlighted incidence of an immune disease involving CD1 in ABL (19)
Cardiac: Premature cardiac death is known in ABL. The exact pathogenesis is uncertain but perhaps related to lipofuscin deposition. (9) Besides, myopathy or neuropathy can lead to cardiomyopathy or respiratory failure. (6) Low platelet and neutrophil response in ABL patients with low LDL levels could be favorable in the context of reducing the ‘pathological' inflammatory/thrombotic responses. (20) Remarkably, the antiatherosclerotic properties of HDL (anti-aggregatory effect on platelets) remain unaffected even after the oxidation of HDL. (15) Atherosclerotic plaques are not a dominant feature in the coronary arteries of ABL patients. Based on this, prevention of atherosclerotic disease by pharmacological inhibition of the MTP gene (lomitapide) (15) to lower plasma cholesterol levels has become a growing concept. (2,15)
Reproductive system: Spontaneous pregnancy can occur despite reduced fertility in ABL. However, in these situations, balancing the vitamin levels is critical. (2)
Malignant potential: Gastrointestinal (7) and neurological malignancies (10) have been diagnosed in ABL patients. Vitamin A and probably vitamin E too potentially have some antineoplastic properties (10,21). Whether chronic vitamin A and E deficiencies could predispose a patient with ABL to a cancerous state and whether extended vitamin supplementation could prevent this complication, is worth exploring further. A possibility of an inverse relation between serum retinol and cancer incidence has been raised (10,22). The relationship between low cholesterol and cancer was also considered and was thought to be a secondary association. (10,22)
Diagnosis:
A high index of suspicion in patients with the characteristic clinical traits (Table:1) is the key. The initial screening test is the serum lipid profile. The ultimate "gold standard" diagnostic test is molecular testing by sequencing the MTP gene. (13) Genetic testing is primarily used for confirmation of the diagnosis where ABL is suspected in a patient. It has no role in predictive testing or prenatal diagnosis. (1)
Differential Diagnosis
Abnormal lipid transference: Autosomal dominant familial hypobetalipoproteinemia (FHBL) is another similar disorder caused by mutations in APO-B gene. (15) Heterozygous FHBL individuals are usually asymptomatic, but may develop fatty liver disease. The homozygous FHBL in general, shares the biochemical and clinical spectrum, and treatment regime of ABL. (15) Differentiating between the two assists in estimating the prognosis. On screening the obligate heterozygote parents of patients with ABL and homozygous FHBL, the lipid profile is normal in the former and reduced to about half of standard levels in the latter. (6) Another difference that was seen on measurement of ApoB-48 and remnant-like particle (RLP) TG levels before and after an oral fat tolerance test in these patients, the fasting and post-oral fat loading levels in homozygous FHBL were higher than those of the patient with ABL. (23) For unclear reasons, patients with HFBL develop liver disease more often than patients with ABL. (1,4) Nevertheless, hepatic involvement in either condition usually has no significant morbid consequence. (4) Chylomicron retention disease (CRD)is another inherited disorder of lipid transference that may show few clinical features of ABL but have relatively higher TG and LDL-C levels. (4) Malabsorption syndromes: Endoscopy findings as discussed above can be the distinctive aspect. (9) Acanthocytosis and CNS: Other neuroacanthocytosis syndromes are X- linked McLeod syndrome (4). In addition, Friedrich’s ataxia and isolated vitamin E deficiency need to be excluded. The abnormal lipid profile can assist in narrowing the diagnosis to either ABL or HHBL. (4) Retinitis Pigmentosa: Whilst retinal disease in ABL presents in the first two decades, the isolated hereditary retinitis pigmentosa, appears later, usually during the second or third decade. (24) Other differentials that can be considered are Ataxia with vitamin E deficiency (AVED), Hypoprebetalipoproteinemia, Acanthocytosis and Pallidal degeneration (HARP syndrome) (13)
Treatment:
There is no definitive cure in ABL. However Many short-term and long-term studies have demonstrated that prompt treatment with high doses of vitamin A and E supplementation can arrest the progression of the neuro-ocular complications. (2,3,4,9,11,15) The cornerstone of management includes dietary modification and high dose fat-soluble vitamin (mainly E and A) administration (Table 2).
Table 2: Management guideline for patients with Abetalipoproteinemia
| THERAPY |
| 1) Dietary modifications |
Low fat diet:
start with 5 grams/day,
grade up as tolerated up to 20 grams/day
MCT supplementation: generally avoided
EFA supplementation: vegetable oils
|
| 2) High dose Fat-soluble vitamins |
Vitamin E:
Dose: 100-300 mg/kg/day
Route: oral (intramuscular also available)
|
Vitamin A:
Dose: 100–400 IU/kg/day
Route: oral
|
Vitamin K:
Dose: 5-35 mg per week
Route: either oral or parenteral
|
Vitamin D:
Dose: 800–1200 IU/day
Route: oral
|
| Other nutrients like Iron and folate |
Recommended Daily Allowance
|
(Above table is based on information adapted from Ref: (4,9,8,6,15)
Dietary changes: Limiting dietary fat stabilizes, the gastrointestinal symptoms and facilitates absorption of other essential nutrients required for maintaining growth and development. (6,16) Dietary fat intake in children can be restricted to 5 grams/day, to begin with; later it can be graded up some more, as tolerated, (9) (limiting the total fat intake up to about 20 grams/day). (25) Polyunsaturated fats are better tolerated than saturated fats. (8) Avoid long-chain saturated fatty acids in particular. (8) Prolonged MCT supplementation is discouraged. (9,8,11) Transient supply of MCT in ABL has been used exclusively during the initial growth spurt, and under vigilant monitoring for hepatotoxicity. (2). This is controversial and preferably avoided if growth velocity is tracking along well. Maintaining adequate caloric intake is crucial. (9)
Essential fatty acids (EFA): EFA are vital nutrients for body function. Notably, docosahexaenoic acid (DHA) maintains nerve and retinal function. (8). EFA are available as vegetable oils containing polyunsaturated fatty acids (e.g., corn or safflower).
Parenteral fat emulsion and vitamins infusions have not received much support given concerns regarding its association with hepatic complications. (4) Its invasive nature also makes it less favourable.
High dose vitamin replacement: Vitamin E: Massive doses up to 100-300 mg/kg/day are used. (4,9 ) The preferred route of administration is oral. An alternative intramuscular route is increasingly being adopted. (3) Serum level of vitamin E is unreliable, and the alternative measurement of vitamin E in adipose tissue (8), is an invasive option. The option of RBC vitamin E (4) or Vitamin E/serum cholesterol ratio (2) can be used to titrate the dose of vitamin E. Fortunately, the risk of toxicity with vitamin E intake is very low. (9) Vitamin A: Doses recommended for children are between (100–400 IU/kg/day).(6,15) Vitamin A dosing can be titrated according to serum carotene concentrations. (8) A risk of vitamin A toxicity does exist. Papilledema was described in a patient with ABL on vitamin A therapy even with serum vitamin A levels within normal limits. (4) Therefore, additional clinical monitoring for vitamin A toxicity with fundoscopy is advised. Vigilant monitoring should be provided in periconceptional females because of the risk of teratogenicity. (2) The dose is halved in this situation. (4)
Vitamin D: Regular replacement with vitamin D is recommended with 800–1200 IU/day in all patients, especially during growth. (4,9,15) Vitamin K: As large doses of vitamin E can potentially worsen the vitamin k deficiency, (9) prophylaxis with vitamin K is recommended. (6,9,) The dose range is 5-35 mg per week by either oral or parenteral route. (4) Other nutrients like iron and folic acid should be administered if a secondary deficiency is identified.
Offering Genetic counseling and the option of prenatal diagnosis will assist the family in planning for future pregnancies. (3)
Follow up:
As clinical spectrum of ABL evolves with time, continued disease surveillance is imperative. The aim is to screen for the progression of existing complications as well as for the appearance of new ones, while simultaneously observing the compliance and adequacy of treatment. Clinical and laboratory monitoring guideline is discussed in Table 3.
Table 3: Follow up outline for patients with Abetalipoproteinemia
| |
Clinical evaluation every 6-12 months |
Annual Laboratory investigations |
| General |
Growth (Height/weight)
Education,
Family participation and
Support
|
LIPIDS* |
Serum levels of:
Total cholesterol
Triglyceride
LDL-cholesterol
HDL-cholesterol
Apo-B
|
| Diet (Dietician reviews) |
Low fat diet
Adequate caloric intake
EFA and Vitamin supplementation
|
|
|
| Gastrointestinal |
Appetite
Diarrhea
Vomiting
Esophagitis
Abdominal distention
Hepatomegaly
|
Hepatic |
AST
ALT
GGT
Total and direct bilirubin
ALP
Albumin
|
| Neurological |
Expected development for age
Ataxia
Dysarthria
Hyporeflexia
Proprioception loss
Muscle pain or weakness
|
Vitamins |
Beta-carotene
25-OH vitamin D
vitamin E/serum cholesterol ratio (reference range >2.2) (2)
INR (
|
| Ophthalmology |
Fundoscopy for papilledema (vitamin A toxicity) |
Other |
CBC
Vitamin B12
Folate
Calcium
Phosphate
Uric acid
Thyroid stimulating hormone
|
| Reproductive |
Vigilant monitoring in periconceptional females as risk of teratogenecity |
|
|
| |
|
Additional Investigations : for age >10 years |
| |
|
Hepatic ultrasonography
Neurological examination
Ophthalmological examination
Bone mineral density via DXA
Echocardiography
|
Every 3 years
Every 6–12months
Every 6–12months
Every 3 years
Every 3 years
|
*After baseline lipid profile, yearly follow-up is not essential as
these levels typically remain stable over the long term.
ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate
aminotransferase; CBC, complete blood count; DXA, dual energy X-ray
absorptiometry; GGT, ?gamma-glutamyl transferase; Reproduced with copyright permission from (4).
Prognosis:
ABL is a lifelong disease with no definitive cure. The prognosis is variable. Untreated patients can culminate in substantially incapacitating disabilities (immobilization and blindness). On the other hand, timely intervention has shown promising results in minimizing the disease morbidity and mortality.
The growth velocity in children improves, however, patients may not reach their expected potential. Pregnancies have been reported with carefully monitored treatment strategies. (2,3,6) A long-term study of ABL patients conducted over three decades, (4) reflected that chronicity was not a noticeable feature of the liver disease in ABL. However, few ABL cases have developed hepatic fibrosis and cirrhosis and undergone liver transplant. (4,11) In one patient requiring a liver transplant for hepatic cirrhosis; the post-transplant serum lipoprotein profile showed increased to normal levels, but the defect in fat absorption persisted given the continued expression of the mutant MTP in the intestine. (6) Patients with ABL with successful treatment have survived to the sixth decade of life. (4)
Prognostic factors: The role of age at the time of diagnosis and commencement of therapy, as a prognostic factor is variable. (1) A genotype and phenotype association is being explored. Vitamin therapy and APOE genotype are proposed factors affecting the severity of the clinical phenotype in ABL. (7) Also, a missense mutation, S590I, in MTP was associated with a milder phenotype and atypical presentation. (1,7) | | | | Conclusion | | The multifaceted pathogenesis, the diverse presentation and treatment response and the persistence of certain abnormalities even after monitored therapy (3) could indicate unfamiliar mechanisms in the complicated molecular process of ABL and the vitamin E metabolism. It may also imply the need for augmenting current treatment protocol and devising strategies to correct the fundamental metabolic defects. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Burnett JR, Bell DA, Hooper AJ, Hegele RA. Clinical utility gene card for: Abetalipoproteinaemia. Eur J Hum Genet. 2012 Aug;20(8). Epub 2012 Feb 29. [CrossRef]
- Ferreira F, Patel V, Matts S. A successful spontaneous pregnancy in abetalipoproteinemia: Amsterdam or the art of vitamin replacement? BMJ Case Rep. 2014;2014.
- Segal S, Sharma S. Ophthaproblem. Vitamin A and vitamin E. Can Fam Physician. 2005; 51(1079):85–86.
- Lee J, Hegele RA. Abetalipoproteinemia and homozygous hypobetalipoproteinemia: a framework for diagnosis and management. Inherit Metab Dis. 2014; 37:333–339. [CrossRef] [PubMed]
- Semba RD. Nutrition and Health. Totawa NJ. In: Handbook of Nutrition and Ophthalmology. Humana Press Inc. 2001: 93-129.
- Zamel R, Khan R, Pollex RL, Hegele RA. Abetalipoproteinemia: two case reports and literature review. Orphanet J Rare Dis. 2008;3:19. [CrossRef] [PubMed]
- Al-Shali K, Wang J, Rosen F, Hegele RA. Ileal adenocarcinoma in a mild phenotype of abetalipoproteinemia. Clin Genet. 2003 Feb;63(2):135-8. [CrossRef] [PubMed]
- Rader DJ, Brewer HB Jr. Abetalipoproteinemia. New insights into lipoprotein assembly and vitamin E metabolism from a rare genetic disease. JAMA. 1993 Aug 18;270(7):865-9. [CrossRef] [PubMed]
- Berriot-Varoqueaux N, Aggerbeck LP, Samson-Bouma M, Wetterau JR. The role of the microsomal triglygeride transfer protein in abetalipoproteinemia. Annu Rev Nutr. 2000;20:663-97. [CrossRef] [PubMed]
- Newman RP, Schaefer EJ, Thomas CB, Oldfield EH. Abetalipoproteinemia and metastatic spinal cord glioblastoma. Arch Neurol. 1984 May;41(5):554-6. [CrossRef] [PubMed]
- Illingworth DR, Connor WE, Miller RG. Abetalipoproteinemia. Report of two cases and review of therapy. Arch Neurol. 1980 Oct;37(10):659-62. [CrossRef] [PubMed]
- Rajajee S, Sathyasekaran M, Shankar J, Dhathathri L, Anandnathan. Importance of screening the peripheral smear. Indian J Pediatr. 2002 Sep;69(9):821-2. [CrossRef] [PubMed]
- Puech B, De Laey JJ, Holder GE. (eds). Inherited Chorioretinal Dystrophies. Springer-Verlag Berlin Heidelberg. 2014. [CrossRef]
- Kornzweig AL. Bassen-Kornzweig syndrome. Present status. J Med Genet. 1970 Sep;7(3):271-6. [CrossRef] [PubMed] [PMC free article]
- Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia. Curr Opin Lipidol. 2014 Jun;25(3):161-8. [CrossRef] [PubMed] [PMC free article]
- Hussain MM, Walsh MT. Abetalipoproteinaemia. Available at URL: http://rarediseases.org/rare-diseases/abetalipoproteinemia. Accessed on 13th February 2016.
- Demircioğlu F, Oren H, Yilmaz S, Arslan N, Gürcü O, Irken G. Abetalipoproteinemia: importance of the peripheral blood smear. Pediatr Blood Cancer. 2005 Aug;45(2):237. [CrossRef] [PubMed]
- Anoop P, Parker-Williams J. Morphological diagnosis of abetalipoproteinemia and the importance of a freshly prepared peripheral smear. Eur J Haematol. 2009 Dec 1;83(6):606. [CrossRef] [PubMed]
- Zeissig S, Dougan SK, Barral DC, Junker Y, Chen Z, Kaser A, et al. Primary deficiency of microsomal triglyceride transfer protein in human abetalipoproteinemia is associated with loss of CD1 function. J Clin Invest. 2010 Aug;120(8):2889-99. [CrossRef] [PubMed] [PMC free article]
- Croft KD, Beilin LJ. Platelet and neutrophil function and eicosanoid release in a subject with abetalipoproteinaemia. Thromb Res. 1993 Feb 15;69(4):333-42. [CrossRef]
- No authors listed. Vitamin A and cancer. Lancet. 1980 Mar 15;1(8168 Pt 1):575-6. [PubMed]
- Kark JD, Smith AH, Switzer BR, et al: Retinol, carotene, and the cancer/cholesterol association. Lancet. 1981 Jun 20;1(8234):1371-2. [CrossRef]
- Kawashiri MA, Tada H, Hashimoto M, Taniyama M, Nakano T, Nakajima K, et al. Extreme Contrast of Postprandial Remnant-Like Particles Formed in Abetalipoproteinemia and Homozygous Familial Hypobetalipoproteinemia. JIMD Rep. 2015;22:85-94. [CrossRef] [PubMed] [PMC free article]
- Merin S, Auerbach E. Retinitis pigmentosa. Surv Ophthalmol. 1976 Mar-Apr;20(5):303-46. [CrossRef]
- Haldeman-Englert C. Bassen-Kornzweig syndrome. MedlinePlus. June 24, 2007; http://www.nlm.nih.gov/medlineplus/ency/article/001666.htm. Accessed 3/5/2009.
- Hirsch E, Simon M, Villemin B, Coumaros D, Warter JM, Jesel M. [Role of the electrophysiologic examination in the diagnosis of Bassen-Kornzweig syndrome]. Neurophysiol Clin. 1988 Sep;18(5):469-75. French. [CrossRef]
DOI: https://doi.org/10.7199/ped.oncall.2016.13
|
| Cite this article as: | | Sonawane R. ABETALIPOPROTEINEMIA. Pediatr Oncall J. 2016;13: 1-8. doi: 10.7199/ped.oncall.2016.13 |
|