|
What is your diagnosis_?
Olcay Sah1, Medya Namdar1, Yakup Sogutlu1, Suat Bicer2.
1Marmara University Faculty of Medicine, Department of Pediatrics, Istanbul, Turkey,
2Yeditepe University Faculty of Medicine, Department of Child Health and Pediatrics, Istanbul, Turkey.
ADDRESS FOR CORRESPONDENCE
Dr Suat Biçer, Associate Professor, Yeditepe University Hospital, Atasehir, Istanbul, Turkey.
Email: suatbicer@yahoo.com, suat.bicer@yeditepe.edu.tr
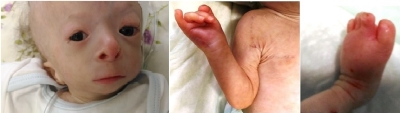
), thin and dry skin, micropenis and small testes. He was second born out of a consanguineous marriage (second degree cousins), and was born by emergency cesarean section at 35 weeks of gestation. Birth weight was 1300 gram. He was operated for anal atresia at 13 day of life. He had recurrent vomiting and diarrhea without a specific diagnosis to date. His sister who was 4.5 years old had the same congenital bone defects plus dermal findings including hyperpigmentation and multiple telangiectasias on her face and extremities.
Figure 1a. Hypotrichosis, saddle nose, low-set ears, micrognathia, absent right thumb, syndactyly, erythematous skin changes, atrophy and telangiectasias.
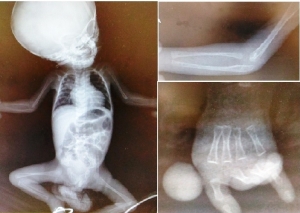
Figure 1b. Radiological findings showing absence of epiphyses, malformed radius, short metacarpal and phalangeal bones, and absence of first metacarpus and phalanx.
Figure 1a
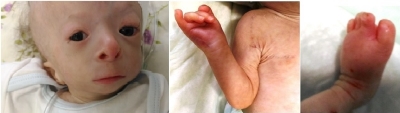
Figure 1b
|
Figure 1a
|
What is the diagnosis?
Rothmund–Thomson syndrome {RTS} or poikiloderma congenitale. It is a rare congenital autosomal recessive disorder that is attributed to mutations of the RECQL4 helicase gene on 8q24. {1} Poikilodermatous skin changes and photosensitivity, skeletal, dental and nail abnormalities, juvenile cataracts, sparse hair, eyelashes, and, or eyebrows, small stature and predisposition to skin cancer and osteosarcoma are some key features of this syndrome. {2,3} Other features in individual cases include gastrointestinal problems in infancy, such as chronic diarrhea and vomiting` hematologic abnormalities including isolated anemia and neutropenia, aplastic anemia, myelodysplasia and leukemia` immune dysfunction` premature aging` sexual abnormalities` cleft palate` micrognathia` anal atresia and sensorineural deafness. {4} RTS has been described in all races and many nationalities and there was no clear gender predilection. {5} No population appears to be at higher or lower risk for the disorder and there are approximately 300 cases described in the literature. {5} The most consistent feature of the syndrome is skin findings. The cheeks are usually first involved with red patches or edematous plaques, sometimes with blistering. Over months to years, the rash enters a chronic stage characterized by atrophy, telangiectasias, pigmentary changes {poikiloderma} and spread to other areas of the face, the extremities, and the buttocks. Skeletal abnormalities of this syndrome are short stature, radial ray anomalies such as absent thumbs, ulnar defects, absent or hypoplastic patella and osteopenia. {5} RTS should be kept in mind with patients who have a small stature and skeletal-dental abnormalities with poikilodermatous skin changes.
Acknowledgements: We are grateful to Hakan Sentürk {writing consultant at the Writing Center, Yeditepe University} for the English revision of this report.
Contributor statement: All authors were involved in the clinical management of the patient, literature search and drafting the document.
Financial disclosure: None
Conflict of interest: None |
| |
| Compliance with ethical standards |
|
Funding: None
|
|
|
Conflict of Interest: None
|
|
- Kitao S, Lindor NM, Shiratori M, Furuichi Y, Shimamoto A. Rothmund-thomson syndrome responsible gene, RECQL4: genomic structure and products. Genomics. 1999;61:268-76. [CrossRef] [PubMed]
- Simon T, Kohlhase J, Wilhelm C, Kochanek M, De Carolis B, Berthold F. Multiple malignant diseases in a patient with Rothmund-Thomson syndrome with RECQL4 mutations: case report and literature review. Am. J. Med. Genet. 2010: 1575-9. [PubMed]
- De Oliveira KM, Silva RA, Carvalho FK, Silva LA, Nelson-Filho P, Queiroz AM. Clinical findings, dental treatment, and improvement in quality of life for a child with Rothmund-Thomson syndrome. Contemp Clin Dent. 2016;7:240–2 [CrossRef] [PubMed] [PMC free article]
- De Somer L, Wouters C, Morren MA, De Vos R, Van Den Oord J, Devriendt K, Meyts I. Granulomatous skin lesions complicating Varicella infection in a patient with Rothmund-Thomson syndrome and immune deficiency: case report. Orphanet J Rare Dis. 2010;5:37. [CrossRef] [PubMed] [PMC free article]
- Wang LL, Plon SE. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al, editors. GeneReviews® (Internet). Seattle (WA): University of Washington, Seattle; 1993-2016. 1999 Oct 6 (updated 2015 Dec3).
|
|
DOI: https://doi.org/10.7199/ped.oncall.2016.46 |
| |
Cite this article as:
Sah O, Namdar M, Sogutlu Y, Bicer S. What is your diagnosis?. Pediatr Oncall J. 2016;13: 112-113. doi: 10.7199/ped.oncall.2016.46
|