Sushant S Mane, Aaditya A Prabhudesai.
Department of Pediatrics, Grant Govt. Medical College, Sir J. J. Group of Hospitals, Mumbai, India.
ADDRESS FOR CORRESPONDENCE
Dr. Sushant S. Mane, Department of Pediatrics, Grant Govt. Medical College, Sir J. J. Group of Hospitals, Byculla, Mumbai 400 008, India.
Email: drsush2006@gmail.com | | Abstract | | The primitive neuroectodermal tumors {PNETs} are rare malignancies usually presenting in the second decade of life with male predilection. They are rarely reported in infancy. We present an eleven months old girl with left thigh soft tissue tumor. Ultrasonography revealed a soft tissue mass in the inter- and intra-muscular plane of the thigh with intact underlying bone suggestive of rhabdomyosarcoma. Tissue biopsy was suggestive of small round cell tumor. Genetic study reported MIC-2 mutation [t(11;22)(q24;q12)] establishing the diagnosis of peripheral PNET. Whole body PET scan revealed metastases in lungs and pelvic bones. Child succumbed to the tumor. | | | | Introduction | | Primitive neuroectodermal tumors (PNETs) are exceedingly rare malignancies, the annual incidence of which is reported to be 2.9 cases per million population from birth upto twenty years of age. (1) These tumors are of neuroectodermal origin belonging to the pathological class of Malignant Small Round Cell Tumors (MSRCT). Peripheral primitive neuroectodermal tumors (pPNETs), which are a subset of PNET, usually present in the second decade of life, with a slight male preponderance. They account for 4-17% of all pediatric soft tissue tumors. (1) These tumors are rare in African American and Asian children, with most cases across the globe occurring in the whites and Hispanic children and adolescents. (1) Though surgical excision and chemotherapy are established treatment modalities for these tumors, the five year survival rate for most cases is less than 25% due to the high incidence of systemic metastases at presentation. (2) We present an 11 months old girl with a left thigh solid tumor. Genetic study reported MIC-2 mutation in tumor cells establishing the diagnosis of peripheral PNET. Whole body PET scan revealed metastases in lungs and pelvic bones. Child succumbed to the tumor. | | | | Case Report | An eleven month old female child, 3rd by birth order, born of non-consanguineous marriage, presented with progressively increasing swelling over the lower half of the left thigh since six months of age. The swelling was painless without any restriction of limb mobility. It was not associated with fever. On examination, the child was playful and healthy with a heart rate of 112 beats/ min and a respiratory rate of 30 breaths/ min. There was no pallor, lymphadenopathy, neurocutaneous markers. Examination of the swelling revealed a mass of 8cm x 8cm x 10cm located on the back of left thigh (Figure 1), which was firm in consistency, non-tender, non-pulsatile, non-transilluminating, with dilated overlying veins without any discharge. Systemic examination was unremarkable. Since the child was asymptomatic and functionally normal the parents delayed seeking medical attention, till the mass grew up to an enormous size. On investigation, hemoglobin was 9.1gm%, white cell count was 13,800/cumm, platelets were 434,000 cells/cumm and erythrocyte sedimentation rate (ESR) was 36 mm/hr. Ultrasonography (USG) of the swelling was suggestive of a large soft tissue mass involving the middle and lower thigh in the intra - and inter - muscular plane with normal underlying bone, likely to be soft tissue sarcoma. Magnetic Resonance Imaging (MRI) showed a 7.5cm x 8cm x 11.3cm well defined, lobulated, homogenously enhancing soft tissue mass in the posterior compartment of the thigh predominantly involving the biceps femoris muscle extending below in the popliteal fossa suggestive of rhabdomyosarcoma along with altered marrow signal intensity of distal femoral diaphysis, which could be representative of marrow metastasis (Figure 2). No evidence of any cortical breach of the bone was noted. The mass was subjected to tissue biopsy. Core needle biopsy was suggestive of malignant small round cell tumor with pPNET as the first differential diagnosis. The cytogenetic study of tumor cells revealed MIC-2 mutation [t(11;22)(q24;q12)]. On immunohistochemistry, tumors cells were strongly positively for CD99 marker which endorsed the diagnosis of pPNET. Positron Emission Tomography (PET) scan showed metastatic foci in lungs, spine and iliac bones. The child was planned for surgical excision and adjuvant chemotherapy but she expired before completion of the treatment.
Figure 1: Lateral View; showing mass lesion in the postero-lateral aspect of the thigh.
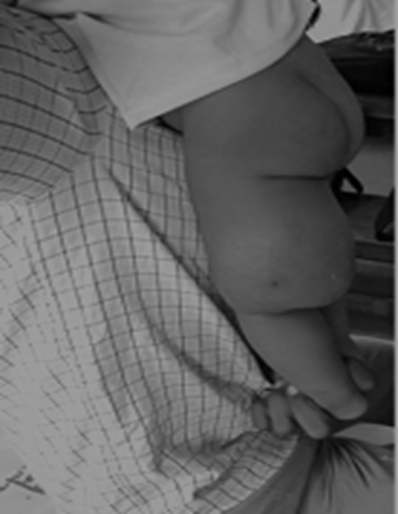
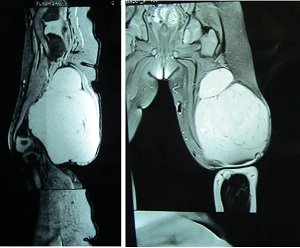
Figure 2: 7.5cm x 8cm x 11.3cm well defined, lobulated, homogenously enhancing soft tissue mass in the posterior compartment of the thigh predominantly involving the biceps femoris muscle extending below in the popliteal fossa suggestive of rhabdomyosarcoma along with altered marrow signal intensity of distal femoral diaphysis.
 | | | | Discussion | Primitive neuroectodermal tumors (PNETs) are a group of highly malignant neoplasms comprising of small round cells, neuroectodermal in origin, affecting soft tissues and bones. PNETs exhibit great diversity in their clinical manifestations but pathologic similarities with other small round cell tumors makes classification of these neoplasm challenging. Batsakis et al (3) divided PNETs into the following three groups based on the tissue of origin: (a) Central Nervous System (CNS) PNETs - Tumors derived from the central nervous system, (b) Neuroblastoma - Tumors derived from the autonomic nervous system, (c) pPNETs - Tumors derived from tissues outside the central and autonomic nervous system. The incidence of pPNETs is likely to be increasing due to the recent diagnostic advances which differentiate these tumors from other small, round cell tumors. pPNETs are also labeled as a part of the Ewing family of tumors (EFTs); these terminologies being often used interchangeably. Ewing’s sarcoma, however, is more common in bones, while peripheral primitive neuroectodermal tumors (pPNETs) are more common in soft tissues. Essentially, they represent different manifestations of the same tumor with similar genetic alterations (MiC-2 mutation, translocation of chromosome 11 and 22) and CD99 positivity. (4,5) The following tumors are classified as pPNETs (6): (a) Ewing sarcoma (osseus and extraosseous), (b) Malignant peripheral primitive neuroectodermal tumors (pPNETs) or peripheral neuroepithelioma of bone and soft tissues, (c) Askin tumor (peripheral neuroepithelioma of the thoracopulmonary region), (d) Other less common tumors (e.g. neuroectodermal tumor, ectomesenchymoma, peripheral medulloepithelioma). Most pPNETs manifest in the thoracopulmonary region (Askin tumor), pelvis, abdomen, and extremities. In a series of cases, reported by Jones and McGill, 11 out of 26 patients had pPNETs in the head and neck presenting as cranial neuropathies, exophthalmos, epistaxis, nasal obstruction, anosmia, neck masses, and headache. (7) Clinical symptoms invariably occur as a result of mass effect of the tumor mass on surrounding structure. In our case the child presented as painless swelling in the thigh which is unusual; also the metastases detected on PET scan were clinically asymptomatic. The most common sites of metastases are lungs, bones and bone marrow which were consistent with our case. In several case series, the incidence of metastases reported was 20-31%, with 5 year survival rates of 0-25%. (2) The prognostic factors of pPNETs include site, tumor volume, and the presence of metastasis. (8)
On histopathology, these tumors comprise of malignant, small, round, relatively undifferentiated cells with scanty basophilic cytoplasm and large hyperchromatic nuclei, referred to as Malignant Small Round Cell Tumors (MSRCT). MSRCT include Ewing's sarcoma (EWS), rhabdomyosarcoma, synovial sarcoma, non-Hodgkin's lymphoma, retinoblastoma, neuroblastoma, hepatoblastoma, nephroblastoma, small cell osteogenic sarcoma, undifferentiated hepatoblastoma, granulocytic sarcoma, and intra-abdominal desmoplastic small round cell tumor. The chromosomal translocation of 11 and 22 differentiates EFT and pPNET from other MSRCT. Rhabdomyosarcomas, lymphomas and neuroblastomas are poorly differentiated in contrast to EFT and pPNET. But the fact remains, histopathology alone cannot be decisive in the diagnosis; with immunohistochemistry and genetic studies being pivotal in confirmation of the tumors. As pPNETs have a high incidence of metastases at presentation, screening with chest radiography, High Resolution CT scan of the thorax, PET scan is advocated in all suspected cases.
Current recommendations advocate complete surgical resection with negative margins whenever possible, adjuvant chemotherapy, if indicated for primary as well as metastatic disease. Carvajal and Meyers, in a comprehensive review of the chemotherapeutic regimens in the treatment of PNETs and Ewing family of tumors (EFTs), recommend a regimen that includes vincristine, doxorubicin, and cyclophosphamide with ifosfamide and etoposide. (8) In some cases, however, the aggressive nature and diffuse spread of these tumors precludes complete surgical excision; in such circumstances palliation remains the only option. As in our case, the Paediatric Oncology sub specialty team had planned surgical excision of the tumor along with adjuvant chemotherapy and supportive treatment. However, the child succumbed before completion of the therapy.
Thus, this case highlights the fact that, PNET should be considered in the differential diagnosis of soft tissue masses even in infantile age group. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Windfuhr JP. Primitive neuroectodermal tumor of the head and neck: incidence, diagnosis, and management. Ann Otol Rhinol Laryngol. 2004;113(7):533-43. [CrossRef]
- Scurr M, Judson I. How to treat the Ewing's family of sarcomas in adult patients. Oncologist. 2006;11(1):65-72. [CrossRef]
- Batsakis JG, Mackay B, el-Naggar AK. Ewing's sarcoma and peripheral primitive neuroectodermal tumor: an interim report. Ann Otol Rhinol Laryngol. 1996;105 (10):838-43. [CrossRef]
- Turc-Carel C, Aurias A, Mugneret F, Lizard S, Sidaner I, Volk C et al. Chromosomes in Ewing's sarcoma. I. An evaluation of 85 cases of remarkable consistency of t(11;22)(q24;q12). Cancer Genet Cytogenet. 1988;32(2):229-38. [CrossRef]
- Marina NM, Etcubanas E, Parham DM, Bowman LC, Green A. Peripheral primitive neuroectodermal tumor (peripheral neuroepithelioma) in children. A review of the St. Jude experience and controversies in diagnosis and management. Cancer. 1989;64(9):1952-60. [CrossRef]
- Castro EC, Parwani AV. Ewing sarcoma/primitive neuroectodermal tumor of the kidney: two unusual presentations of a rare tumor. Case Report Med. 2012;2012:190581.
- Jones JE, McGill T. Peripheral primitive neuroectodermal tumors of the head and neck. Arch Otolaryngol Head Neck Surg. 1995;121(12):1392-5. [CrossRef]
- Carvajal R, Meyers P. Ewing's sarcoma and primitive neuroectodermal family of tumors. Hematol Oncol Clin North Am. 2005;19(3):501-25, vi-vii. [CrossRef]
DOI: https://doi.org/10.7199/ped.oncall.2018.21
|
| Cite this article as: | | Mane S S, Prabhudesai A A. Primitive Neuroectodermal Tumor in Infancy - An Unusual Clinical Presentation.. Pediatr Oncall J. 2018;15: 43-45. doi: 10.7199/ped.oncall.2018.21 |
|