Is it Ehlers Danlos syndrome or Osteogenesis Imperfecta?
|
|
Is it Ehlers Danlos syndrome or Osteogenesis Imperfecta?
07/02/2019
07/02/2019
Devi Bavishi*, Ira Shah**, Ashok Johari***
https://www.pediatriconcall.com/Journal/images/journal_cover.jpg
Devi Bavishi1, Ira Shah2, Ashok Johari3.
1Seth G S Medical College, Mumbai, India,
2Department of Pediatrics, Levioza Health Care, Mumbai, India,
3Department of Paediatric Orthopaedics and Spine Surgery, Children's Orthopedic Centre, Mumbai, India.
ADDRESS FOR CORRESPONDENCE
Devi Bavishi, Seth G S Medical College, Mumbai, India.
Email: devi_bavishi@yahoo.co.in
Fractures, EDS, OI
|
Clinical Problem
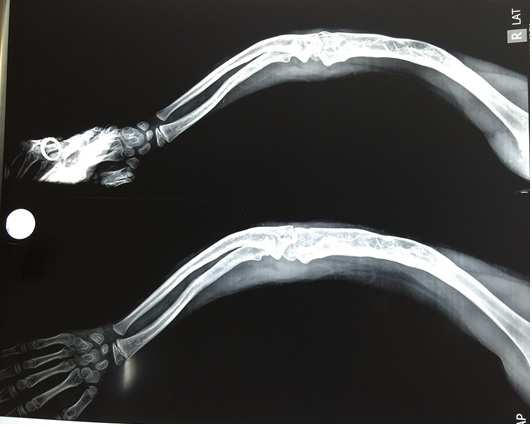
A two and a half years old female, born of a non-consanguineous marriage presented in August 2011 with multiple fractures affecting all limbs since the age of nine months. She had 36 fractures till date and her most recent fracture was in July 2011. She had been on bisphosphonates (initially pamidronate 12 mg/kg every two months) for the same, but fractures continued. She had received zoledronate in December 2010 following which she developed redness in both eyes which were detected to be endophthalmitis, which gradually progressed to complete loss of vision and retinal detachment in both eyes. There were no similar complaints in other family members. Her mental development was normal but she could not walk due to deformities. Her serial bone mineral densities showed osteopenia. On presentation to us in August 2011, she had deformed bones (knock knees, incurving of both hands and right leg with medial rotation) and bilateral eye phthisis. She had no joint laxity or hearing problems. Her X-ray of the right forearm showed severe incurving of the radius and ulna bones (Figure 1). Based on the clinical presentation, she was clinically suspected to have osteogenesis imperfecta (OI). She was put on vitamin D3 and calcium supplements. After two months, there were no new fractures and she could walk. As her parents were planning to have another child, a molecular test was done which showed heterozygous status for p. (Arg46Cys) (NM_000302.3:c.136C>T) mutation in Exon 2 and p.P692L (NM_000302.3:c.2075C>T) mutation in Exon 19 of PLOD1 gene suggestive of autosomal recessive Ehlers-Danlos Syndrome Type VI.
|
Figure 1. X-ray of the right forearm showing severe incurving.
|
| |
Is this osteogenesis imperfecta or Ehlers-Danlos Syndrome type VI?
|
|
|
Discussion
Ehlers Danlos Syndrome (EDS) is a heterogeneous group of connective tissue disorders resulting in varying degrees of skin fragility, skin hyperextensibility and joint hypermobility due to a defect in the structure, synthesis, or processing of collagen or biomolecules that interact with collagen.1,2 There are many different types of EDS which are classified according to the new The 2017 International Classification of the Ehlers-Danlos Syndromes.3 kEDS-PLOD1 (Kyphoscoliotic type) is an autosomal recessive disorder, its incidence being 1; 100,000 live births4 which are caused due to mutations in the PLOD gene which encodes a collagen-modifying enzyme known as lysyl hydroxylase.4,5 The disorder is characterized at birth by severe muscular hypotonia, kyphoscoliosis, marked joint hypermobility and subluxations, severe skin hyperelasticity, the fragility of the skin with abnormal scarring, osteopenia (without a tendency to fractures).6 The sclera may be unusually fragile, to which even minor trauma may result in rupture of the sclera, cornea, and/or detachment of retina.5 Our patient had blue sclera and retinal detachment in both eyes leading to vision loss and phthisis. However, our patient also had recurrent fractures which have not been described previously in kEDS-PLOD1. She also did not have skin hyperelasticity.
The major inherited cause of multiple fractures in childhood is the condition called Osteogenesis Imperfecta (OI). The features of OI are osteoporosis, fragile bones that fracture easily, ligament laxity and hypermobility of the joints. The estimated incidence is approximately 1/100,000 to 1/25,000.7 Retinal detachment has also been described in OI.8
Confirmatory molecular testing of kEDS-PLOD1 is dependent on the identification of homozygosity or compound heterozygosity for pathogenic variants in the PLOD1 gene that lead to a deficiency of the collagen-modifying enzyme Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 (PLOD1 or LH1 [lysylhydroxylase1]).4 Since the parents were not tested, it is not known if there is compound heterozygosity.
Thus, based even on the genetic report, it is not possible to confirm the diagnosis of ED type 6 in this child. It is always important to determine whether the heterozygous mutation is the cause of the disease by doing the genetic test in the parents.
|
| |
| Compliance with ethical standards |
|
Funding: None
|
|
|
Conflict of Interest: None
|
- Shipley M, Rahman A, O'Gradaigh D, Compston J. Skeletal dysplasia. In: Kumar P, Clark M, (eds). Kumar & Clark's Clinical Medicine. 7th ed. Edinburgh. Saunders Elsevier. 2009:568
- Mao J, Bristow J. The Ehlers-Danlos syndrome: on beyond collagens. J Clin Invest. 2001; 107: 1063–1069. [CrossRef] [PubMed] [PMC free article]
- Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175:8-26. [CrossRef] [PubMed]
- Yeowell HN, Steinmann B. PLOD1-Related Kyphoscoliotic Ehlers-Danlos Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al. (eds). GeneReviews [Internet]. Seattle (WA). University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1462/. Accessed on 26 December 2017.
- Ehlers Danlos Syndrome - NORD (National Organization for Rare Disorders) (Internet). NORD (National Organization for Rare Disorders). Available at: http://rarediseases.org/rare-diseases/ehlers-danlos-syndrome/. Accessed 22 June 2016
- Rohrbach M, Vandersteen A, Yis U, Serdaroglu G, Ataman E, Chopra M, et al. Phenotypic variability of the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VIA): clinical, molecular and biochemical delineation. Orphanet J Rare Dis. 2011;6:46. [CrossRef] [PubMed] [PMC free article]
- Basel D Steiner R. Osteogenesis imperfecta: Recent findings shed new light on this once well-understood condition. Genet Med. 2009;11:375-85 [CrossRef] [PubMed]
- Fleissig E, Barak A. Surgical management of retinal detachment in osteogenesis imperfecta: case report and review of the literature. Retin cases brief rep. 2019;13:43-46. [CrossRef] [PubMed]
|
|
| |
Cite this article as:
Bavishi D, Shah I, Johari A. Is it Ehlers Danlos syndrome or Osteogenesis Imperfecta?. Pediatr Oncall J. 2019;16: 133-134. doi: 10.7199/ped.oncall.2019.62
|