Rajesh K Kulkarni, Aarti A Kinikar, Brijender Prasad, Greeshma Nair, Sagar Vartak.
Department of Pediatrics, B J Government Medical College, Pune, Maharashtra, India.
ADDRESS FOR CORRESPONDENCE
Dr Aarti Kinikar, Department of Pediatrics, B J Government Medical College, Pune, Maharashtra, India.
Email: aarti.kinikar63@gmail.com | | Abstract | | Hyaline fibromatosis syndrome is a very rare condition caused by mutations in the ANTXR2 gene and characterized by nodular skin lesions, joint contractures and visceral involvement in the severe variant. Most children who have the severe variant (Infantile systemic hyalinosis-ISH) succumb by 2 years of age while those with milder form (Juvenile Hyaline Fibromatosis-JHF) without visceral involvement may live till second decade. We present an 8 years old boy with overlap features of ISH and JHF and a dysplastic kidney. Association of dysplastic kidney with HFS has not been reported before to the best of our knowledge. | | | | Keywords | | hyaline fibromatosis syndrome, ANTXR2 gene | | | | Introduction | | Hyaline fibromatosis syndrome (HFS) is a very rare condition characterized by deposits of hyaline in the skin and in various other body tissues.1,2 Previously infantile systemic hyalinosis (ISH) and Juvenile hyaline fibromatosis (JHF) were considered distinct entities, however now they are thought to represent severe and mild manifestations of the same gene defect.3 Less than 70 cases of JHF and 20 cases of ISH have been reported worldwide.4 Majority of the cases have been reported in the Arab population with only a few cases reported from India.5 The clinical features common to both conditions include joint contractures, papular and nodular skin lesions, gingival hypertrophy, osteopenia, and normal brain development. Common distinguishing features of ISH include thickened skin, erythema, or hyperpigmentation over bony prominences, visceral involvement, persistent diarrhea, frequent severe infections, failure to thrive and death usually by 2 years of age. We present an 8 years old boy with overlapping features of both ISH and JHF along with dysplastic kidney. | | | | Case Report | An 8 years-old boy, born of third degree consanguineous marriage presented with diarrhea for 7 days and inability to put on weight. There was history of one intrauterine death at 8 months gestation and one spontaneous abortion at 8 weeks of gestation in the previous pregnancies. This child was born by normal vaginal delivery and had a birth weight of 3 kg. At 4 months age, he developed restricted movement of limbs and skin lesions in the form of multiple nodules over scalp and abdomen which progressed to involve the nose, face, ears, scalp and perianal regions. By one year of age, he gradually developed severe joint contractures in most of the large joints. He had delayed motor milestones. Over the last 7 years, baby suffered from multiple episodes of diarrhea, recurrent respiratory infections and had poor weight gain. On presentation, the child was severely dehydrated, weighed 16 kg (<5th centile) with height of 93 cm (<5th centile) with blood pressure of 80/46 mmHg. There was severe muscle wasting and edema with flexion joint contractures at both elbow and knee joints. Skin over all the limbs was indurated and there were multiple nodular lesions on knuckles, knees, ankles (Figure 1). Scalp showed greasy scales and mild erythema and multiple large nodules (Figure 2). There were multiple, pearly, pink, grouped papules of variable sizes over the bridge of the nose, nasolabial folds, perioral areas and ears. Several fleshy papular and nodular lesions in the gluteal cleft and perianal region were seen (Figure 3). There were hyperpigmented, subcutaneous plaques over the elbow joints and malleoli. Severe gingival hypertrophy was present (Figure 4). Laboratory studies revealed hemoglobin 6.3 g/dl, total leukocyte count 4800/cumm (neutrophils 58%, lymphocytes 34%, eosinophils 4%, monocytes 4%) with platelet count 4,10,000/cumm. Liver function tests, renal function tests and thyroid profile were normal. Radiographs of long bones showed severe osteopenia. Abdominal ultrasound showed left cystic dysplastic kidney (left kidney size 9.7 cm, right kidney size 8.2 cm) and MRI brain was normal. Skin biopsy from a papulonodular lesion showed homogenous, hyalinized papillary dermis with cellular proliferation of spindle and round cells (Figure 5). Periodic acid Schiff (PAS) staining showed eosinophilic material occupying the deeper dermis. The deposited material did not stain with either Alcian blue or Masson's trichrome. Molecular genetic analysis of targeted mutation in CMG2 (ANTXR2) gene using PCR followed by DNA sequencing confirmed it to be homozygous for c.1153G>A mutation (p.G384S) suggestive of HFS.
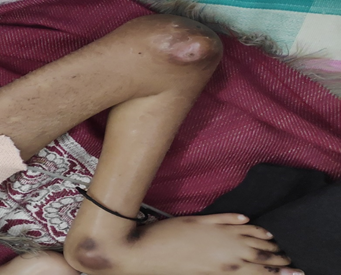
Figure 1. Multiple hyperpigmented nodular lesions on knees and ankles
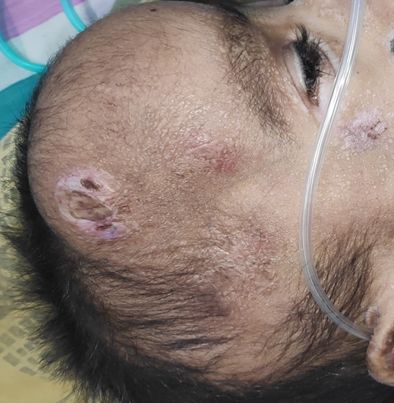
 | Figure 2. Scalp shows multiple large nodules.
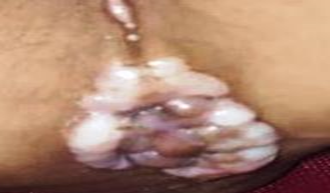
 | Figure 3. Fleshy papular and nodular lesions in the gluteal cleft and perianal region
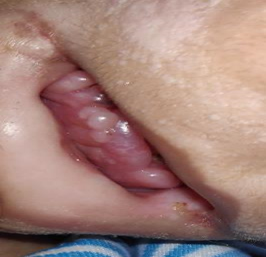
 | Figure 4. Severe gingival hypertrophy
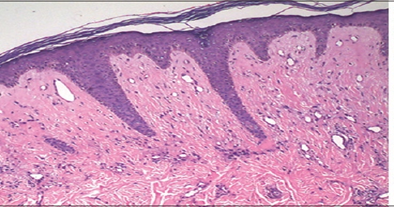
 | Figure 5. Histopathology shows homogenous, hyalinized papillary dermis with cellular proliferation of spindle and round cells
 |
| | | | Discussion | HFS (OMIM 228600) is a rare autosomal recessive condition caused by homozygous or compound heterozygous mutation in the gene encoding capillary morphogenesis protein-2 (CMG2 or ANTXR2) on chromosome 4q21. CMG2 is an integrin-like cell surface receptor that binds laminins and Type IV collagen via a von Willebrand factor Type A domain and in the skin it is thought to play a role in cell-matrix interactions and basement membrane integrity and endothelial cell morphogenesis. Genotype-phenotype studies suggested that missense and other in-frame mutations that affect the cytoplasmic domain tend to cause JHF, whereas truncating mutations and missense mutations that affect the extracellular protein-binding domain tend to cause ISH.1,3
Patients are usually normal at birth. Abnormalities begin in the first few months of life with progressive flexor joint contractures resulting in inability to stand and walk. Characteristic skin lesions are papulonodular skin lesions, gingival hypertrophy, and thickened hyperpigmented macules/patches over bony prominences of the joints.6 Radiological examination of the long bones may show delayed skeletal maturation, severe osteopenia, bony erosions, and lucent defects. ISH is distinguished from JHF by hyaline deposits in multiple organs, recurrent infections, protein-losing enteropathy causing diarrhea, failure to thrive, and death within the first 3 years of life, recurrent chest infections being the leading cause of death.7 Our patient had features similar to both ISH and JHF and a left dysplastic kidney which has not been reported previously. Cognitive development is normal. Clinical differentials include congenital generalized fibromatosis, Winchester disease, Faber's disease, nodular amyloidosis, mucopolysaccharidosis and neurofibromatosis.8 Histopathological examination of a typical papulonodular skin lesion shows deposition of an amorphous hyaline, eosinophilic substance in which spindle-shaped cells are embedded. It may be vaguely chondroid. The material is pas positive and diastase resistant and does not stain with alcian blue.9 Skin biopsy in our patient had hyalinized papillary dermis with cellular proliferation of spindle and round cells suggestive of HFS. Genetic testing for mutation in ANTXR2 on chromosome 4q21 is also available. Similarly our patient was homozygous for CMG2 (ANTXR2) mutation.
Treatment is supportive. penicillamine has been tried with limited success.10 Early surgical excision is recommended by few experts to prevent new lesions in JHF, however there may be frequent recurrences. Genetic counseling is very important as there is a 25% risk of disease in each pregnancy.
| | | | Acknowledgement | | We acknowledge help received from Dr Koumudi Godbole, Consultant Clinical Geneticist, Deenanath Mangeshkar Hospital, Pune towards the genetic testing of this case. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Vasani R, Parikh D. Hyaline fibromatoses syndrome: A rare entity. Indian J Paediatr Dermatol 2019;20:64-67 [CrossRef]
- Park K, Chang D, Sung M. Juvenile hyaline fibromatosis. Clin Exp Otrhinolaryngol. 2010;3:102-106 [CrossRef] [PubMed] [PMC free article]
- Hanks S, Adams S, Douglas J, Arbour L, Atherton D, Balci S. Mutations in the Gene Encoding Capillary Morphogenesis Protein 2 Cause Juvenile Hyaline Fibromatosis and Infantile Systemic Hyalinosis. Am J Hum Genet. 2003;73:791–800 [CrossRef] [PubMed] [PMC free article]
- Al Mayouf SM, AlMehaidib A, Bahabri S, Shabib S, Sakati N, Teebi AS, et al. Infantile systemic hyalinosis: A fatal disorder commonly diagnosed among arabs. Clin Exp Rheumatol. 2005;23:717 720 [PubMed]
- Shah I. Dermal swellings, joint contractures but no gingival hypertrophy. Pediatr Oncall J. 2017;14:100. [CrossRef]
- Krishnamurthy J, Dalal BS, Sunila, Gubanna MV. Juvenile hyaline fibromatosis. Indian J Dermatol. 2011;56:731–733. [CrossRef] [PubMed] [PMC free article]
- Mancini GM, Stojanov L, Willemsen R, Kleijer WJ, Huijmans JG, van Diggelen OP, et al. Juvenile hyaline fibromatosis: Clinical heterogeneity in three patients. Dermatology 1999;198:18 25 [CrossRef] [PubMed]
- Coffin C, Alaggio R. Pediatric Spindle Cell Tumors. In: Hornick J (ed). Practical Soft Tissue Pathology: a Diagnostic Approach. 2nd edn. Elsevier. 2018: 101-134
- Momin YA, Bharambe BM, D'Costa G. Juvenile hyaline fibromatosis: A rare lesion. Indian J Pathol Microbiol. 2011;54:838 839 [PubMed]
- Shieh JT, Swidler P, Martignetti JA, Ramirez MC, Balboni I, Kaplan J, Kennedy J, et al. Systemic Hyalinosis: A Distinctive Early Childhood–Onset Disorder Characterized by Mutations in the Anthrax Toxin Receptor 2 Gene (ANTRX2). Pediatrics. 2006;118:1485-1492. [CrossRef] [PubMed]
DOI: https://doi.org/10.7199/ped.oncall.2019.47
|
| Cite this article as: | | Kulkarni R K, Kinikar A A, Prasad B, Nair G, Vartak S. Hyaline Fibromatosis Syndrome: Report and Literature review of a Rare and Fatal Genetic Disorder. Pediatr Oncall J. 2019;16: 83-85. doi: 10.7199/ped.oncall.2019.47 |
|