D Enyama1,2, C A Ngo Kana3, J Mayouego Kouam4, D Noukeu Njinkui1,2, F Kemta Lekpa2,5, P C Mbonda6,7, D C Kedy8, E Mbonda6, S Nguefack3,6.
1Department of Pediatrics, Douala Gynaeco-Obstetric and Pediatric Hospital, Douala, Cameroon,
2Faculty of Medicine and Pharmaceutical Sciences, University of Dschang, Cameroon,
3Department of Pediatrics, Yaoundé Gynaeco-Obstetric and Pediatric Hospital, Yaoundé, Cameroon,
4Department of Ophthalmology, Douala Laquintinie Hospital, Douala, Cameroon,
5Department of Internal Medicine, Douala General Hospital, Douala, Cameroon,
6Faculty of Medicine and Biomedical Sciences, University of Yaoundé I, Cameroon,
7Department of Internal Medicine, Yaoundé General Hospital, Yaoundé, Cameroon,
8Faculty of Medicine and Pharmaceutical Sciences, University of Douala, Cameroon.
ADDRESS FOR CORRESPONDENCE
Dominique ENYAMA, Faculty of Medicine and Pharmaceutical Sciences University of Dschang, P O Box 96, Dschang, Cameroon.
Email: enyamad@yahoo.fr | | Keywords | | ataxia, telangiectasia, children, Cameroon | | | | Abstract | Background: Ataxia-Telangiectasia (A-T) is a rare genetic disease characterized by progressive ataxia and multisystem involvement, which requires early diagnosis and multidisciplinary management.
Methods: We report a series of seven patients with A-T. This is a retrospective study conducted from 2016 to 2020, in patients with a history of progressive cerebellar ataxia and ocular telangiectasia on physical examination as well as elevated serum alpha-fetoprotein. Data were collected from the medical records of the patients.
Results: There were three boys and four girls. The mean age at onset of symptoms was 36.4 months (range 12 to 72 months) and the mean age at diagnosis was 84 months (range 60 to 144 months). Family history of A-T was noted in 3 patients. There was no parental consanguinity in any patient. Ataxia was the first sign of the disease and the reason for consultation in all patients. Cerebellar ataxia and ocular telangiectasia were seen in all patients. Malnutrition was present in 6 (85.7%) patients, dysarthria was observed in 3 (42.8%) patients, intention tremors in 2 (28.6%) patients, dystonia and epilepsy in 1 (14.3%) patient each. Recurrent infections with repeated hospitalizations was present in 1 (14.3%) patient. Elevated serum alpha-fetoprotein level was seen in all patients with a mean level of 311.39 ng/ml (range from 113 ng/ml to 911.6 ng/ml). Four (57%) patients underwent brain imaging which showed cerebellar atrophy in three of them. Genetic testing for A-T was not done in any patient due to unavailability. All these patients were still alive when we submitted this paper. A motor deficiency worsening was observed in 2 patients, who currently use a wheelchair since the age of 6 and 7 years old, respectively.
Conclusion: In children developing gait disorders with progressive cerebellar ataxia, physicians should look for ocular telangiectasia. In our setting, the diagnostic confirmation is based on elevated serum alpha-fetoprotein level.
| | | | Introduction | | Ataxia-Telangiectasia (A-T) is an autosomal recessive disease characterized by a progressive cerebellar ataxia, oculocutaneous telangiectasia and recurrent sinopulmonary infections.1 In addition, A-T is also characterized by immunodeficiency, radiation sensitivity, premature aging, and a predisposition to cancer development, especially of lymphoid origin. Other abnormalities include poor growth, gonadal atrophy, delayed pubertal development and insulin resistant diabetes.1 It is caused by mutations in Ataxia-Telangiectasia Mutated (ATM) gene encoding a serine/threonine protein kinase.2,3 A-T is reported all over the world with an estimated prevalence varying between 1:40 000 and 1:100 000.4,5,6 Diagnosis of A-T is usually made by the combination of clinical features and a variety of paraclinical abnormalities, including elevated and slowly increasing serum alpha-fetoprotein (AFP) levels after two years of age and cerebellar atrophy detected on neuroimaging.1 The diagnosis can be confirmed by the absence or deficiency of ATM protein and/or ATM kinase activity in cultured cell lines established from lymphocytes or skin biopsies4,6,7,8 or the identification of pathological mutations in the ATM gene.7,8 In Sub-Saharan Africa in general and particularly in Cameroon, data on Ataxia-Telangiectasia in children are scarce. For that reason, we conducted this study to determine the clinical profile of children presenting with Ataxia-Telangiectasia in Cameroon. | | | | Methods & Materials | | This study reports a case series of patients who consulted at the child neurology clinic in two university teaching hospitals in Cameroon (Douala Gynaeco-Obstetric and Pediatric Hospital and Yaoundé Gynaeco-Obstetric and Pediatric Hospital). It was a retrospective and descriptive study, carried out from March 2016 to March 2020. Data were collected from patients’ medical records. We included 7 patients with a suspicion of A-T according to these three following criteria: progressive cerebellar ataxia, ocular telangiectasia and elevated serum alpha-fetoprotein. One patient who presented with progressive cerebellar ataxia with ocular telangiectasia but who did not undergo serum alpha-fetoprotein dosage was not included in the study. We analyzed socio-demographic data (age, sex), family history of ataxia, consanguinity, age at onset of first symptoms, age at diagnosis, clinical signs (ataxia, telangiectasia, oculomotor apraxia, dystonia, tremors, epilepsy, dysarthria), weight growth, recurrence of infections, serum alpha-fetoprotein level, brain imaging results, management and course. We used World health Organization (WHO) anthropometric software version 3.2 for the analysis of physical development of the children. | | | | Results | A total of seven patients were involved in the study, including three boys and four girls. Family history of A-T was noted in three patients among whom two patients were siblings. Parental consanguinity was not found in any case. The age at onset of first symptoms ranged from 12 to 72 months with a mean age at onset of 36.4 months. Age at diagnosis ranged from 60 to 144 months with a mean age at diagnosis of 84 months. On clinical examination, ataxia was the first sign of the disease and the presenting complain in all patients. In one patient, psychomotor regression was associated with ataxia as initial symptoms of the disease. Ataxia and telangiectasia were seen in all patients (Figure 1). Several ocular abnormalities were noted in one patient and these included ocular telangiectasia, oculomotor apraxia, oculomotor palsy and ptosis. Malnutrition was present in 6 (85.7%) patients, dysarthria was observed in 3 (42.8%) patients, intention tremors in 2 (28.6%) patients, dystonia and epilepsy in 1 (14.3%) patient each. Recurrent infections with repeated hospitalizations were present in 1 (14.3%) patient. All these clinical features are summarized in Table 1.
Table 1. History, clinical and paraclinical description of all patients
| Patient |
Gender |
Age (years) |
Age at first symptoms (months) |
Age at diagnosis (months) |
Diagnostic delay (months) |
Family history of ataxia |
Clinical signs |
Head circumference (cm) |
BMI
(Z -score) |
Indices W/H
(Z score) |
Indices WA
(Z score) |
Repeated infections |
Cerebellar atrophy |
a-fetoprotein (ng/ml) |
| Patient 1 |
M |
5 |
24 |
60 |
36 |
Yes |
static and kinetic ataxia, telangiectasias |
- |
0.675 |
0.631 |
-1.294 |
No |
- |
113.17 |
| Patient 2 |
F |
7 |
15 |
84 |
69 |
Yes |
static and kinetic ataxia, telangiectasias |
53 |
-1.398 |
- |
-0.196 |
Yes |
- |
209.56 |
| Patient 3 |
F |
7 |
12 |
84 |
72 |
Yes |
static and kinetic ataxia, telangiectasia, dysarthria, tremor, dystonia |
- |
- |
- |
- |
Yes |
- |
145.76 |
| Patient 4 |
M |
7 |
60 |
84 |
24 |
No |
static and kinetic ataxia, telangiectasias, oculomotor apraxia |
49.5 |
-8.788 |
- |
3.756 |
- |
Yes |
491.09 |
| Patient 5 |
M |
6 |
36 |
72 |
36 |
No |
static and kinetic ataxia, telangiectasias, oculomotor apraxia, dysarthria, epilepsy, gross motor impairment, sphincter disorders |
51.5 |
-1.502 |
- |
-0.989 |
Yes |
Yes |
180.54 |
| Patient 6 |
F |
8 |
36 |
96 |
60 |
No |
static and kinetic ataxia, telangiectasias |
50 |
-0.673 |
- |
-1.963 |
No |
No |
911.6 |
| Patient 7 |
F |
12 |
72 |
144 |
72 |
No |
static and kinetic ataxia, telangiectasia, dysarthria, tremors, gross motor impairment |
55 |
0.362 |
- |
-4.747 |
No |
Yes |
128.04 |
The biological features included a high level of serum alpha-fetoprotein in all cases, with concentration values ranging from 113.17 ng/ml to 911.6 ng/ml and a mean concentration of 311.39 ng/ml (normal value <10 ng/ml). These biological features are shown in Table 1.
Magnetic resonance imaging (MRI) was performed in four patients (57%) which showed cerebellar atrophy in three of them (Figure 2). Three patients did not undergo brain imaging.
No patient was able to afford genetic tests, including testing for the ATM gene mutation.
Regarding the disease outcome, we did not record any deaths among our patients during the study period. Motor worsening was observed in 2 patients, who moved to a wheelchair at the age of 6 and 7 years old, respectively. All patients had psychomotor care.
Figure 1. Ocular telangiectasia.
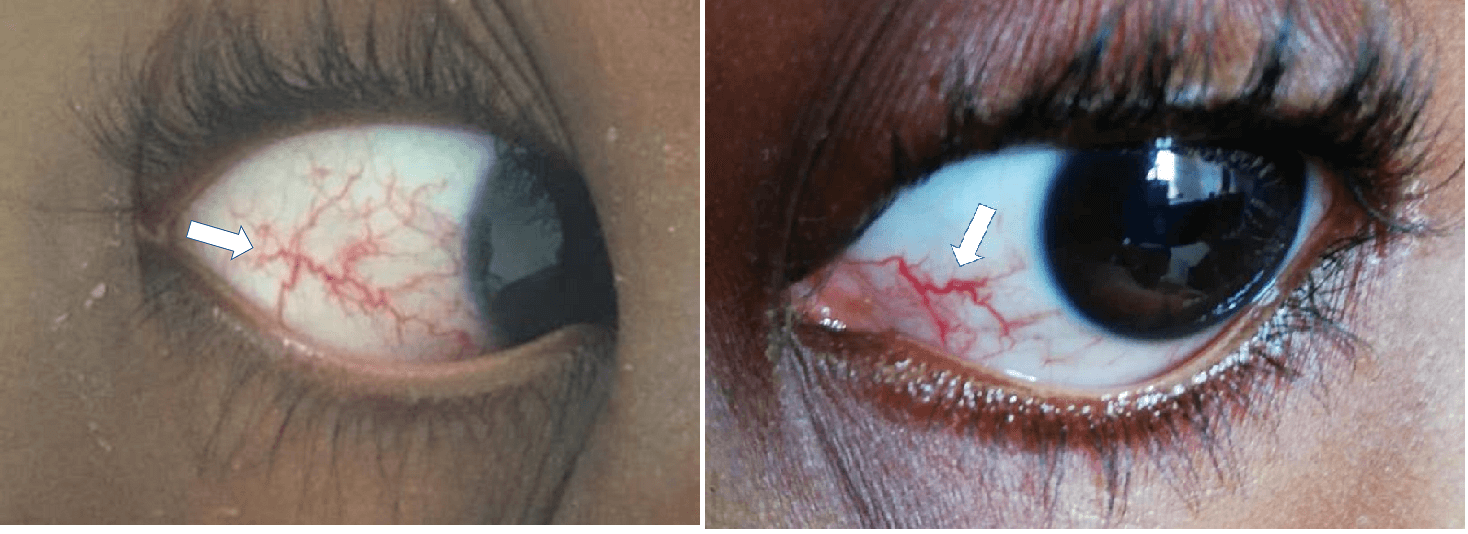
Figure 2. MRI showing cerebellar atrophy (vermis and hemispheres).
.png)
| | | | Discussion | The small number of cases of the disease in our setting is in accordance with the literature which estimates a prevalence between 1/40,000 and 1/100,000.1 Typically, in the natural history of A-T, there are no clinical signs at birth and the disease usually begins around the age of one or two years, when gait is acquired.9 In our study, the mean age at onset of symptoms was 36 months (3 years) and the mean age at diagnosis was 84 months (7 years). Alyasin et al in Iran, observed a mean age at onset of symptoms of 2.56 x 2.55 years and a mean age at diagnosis ranging from 5 to 16 years.10 In our series, delay in diagnostic can be explained by a couple of factors. First of all, the accessibility to a child neurology consultation is difficult for most patients. Then, the low incidence of the disease can make its diagnosis difficult. Lastly, the slow progression of symptoms may lead to misdiagnose the disease in its initial phase.
As one of the main clinical features, ataxia was observed in all patients in our series. It was the first symptom of the disease and the most frequent presenting complain. These results are similar to those of other authors.10,11,12,13 According to many authors, cerebellar ataxia dominates early in the disease, with very unstable walking. Ataxia is first static and then becomes kinetic. Then comes dysarthria, tremors of the extremities and oculomotor apraxia.9
Ocular telangiectasia was also observed in all patients in our series. This can be explained by the mean age of the patients at the time of diagnosis which was 7 years. According to most authors, ocular telangiectasia appear between 5 and 8 years of age.1
One in seven patients had a history of repeated severe infections, which could suggest an immunodeficiency though no specific biological investigation was performed to confirm the immunodeficiency. About two-thirds of people with A-T have abnormalities of the immune system.1 Immunodeficiency relates to the decrease in immunoglobulins A, E, G subclasses and lymphopenia.14 This deficiency does not progress and some studies suggest a normalization of lymphocytes level (T lymphocytes) by the age of 20 years old.14,15,16
Growth retardation was identified in six patients. Growth failure is frequently reported in children with A-T.1,17,18 It is linked to dysphagia and swallowing disorders which usually appear in the second decade, caused by neurological changes that interfere with the coordination of the mouth and pharynx, necessary for effective swallowing.17 This will cause insufficient calorie intake and compromise the patient's growth. Feeding by nasogastric tube or by gastrostomy tube are therefore recommended depending on the severity of the malnutrition, the frequency of inhalation accidents and the impact of the duration of the meals on the patient's daily activities.1
Dysarthria, oculomotor apraxia and tremors observed in some patients in our series have been reported by several studies.10,13,19
Epilepsy was diagnosed in one patient in our series. In a study conducted in Turkey on 91 patients with A-T, Akturk et al observed recurrent convulsions in 3 patients (3.2%) while Alyasin et al recorded 16.7% of cases of epilepsy.10,20 Epilepsy is not found in the typical form of A-T. Some diseases may mimic symptoms and signs of A-T, hence the benefit of genetic testing to avoid misdiagnosis.
Serum alpha-fetoprotein level was high in all patients. This finding is consistent with the literature, which indicates that this level is greater than 10 ng/ml in more than 95% of patients with A-T.1 Levels of AFP appear to increase over time.21 Why the majority of individuals with A-T have elevated levels of AFP remains unknown.1 The serum levels of AFP are lower in patients with A-T than with tumors derived from yolk sac and liver cancers but similar to the levels observed in hepatitis.6 Brain MRI in A-T usually shows cerebellar atrophy of vermis and hemispheres22,23 as was seen in our patients. Genetic tests were not carried out due to unavailability in our milieu. International collaboration with genetics laboratories can help to draw up the genetic profile of these black African patients who could possibly show different genotypes from the ones currently identified in Caucasian populations.
There is currently no specific treatment for A-T. Management is symptomatic and supportive including physical, occupational and speech therapies.1 Psychological support of the patients and their families is also useful. In our series, all the patients benefited from psychomotor therapy associated with regular follow-up in child neurology clinic. Occupational therapy was not available in our context.
Regarding the natural history of the disease, we observed in two patients a progressive deterioration of motor skills requiring assistance in a wheelchair at the ages of 6 and 7 years old respectively. These two patients were seen in child neurology consultation for the first time with already established gross motor dysfunction. This finding is not consistent with the observations of many authors, who noted that the majority of children require wheelchairs assistance from the age of 10 years.6 This early installation in a wheelchair in our series, may be explained by late diagnosis associated with limited care due to the expensive cost of treatment for families or unavailability of certain therapies. No death was recorded in our series. A study reports a median survival age of 25 years with advances in management.17
The present study has some limitations. Due to its retrospective nature, data collection has occasionally been hampered by missing data in patients' medical records. Also, we were unable to carry out genetic studies in our patients because of their unavailability in Cameroon and their high cost.
| | | | Conclusion | | Ataxia-Telangiectasia is a rare disease, with variable clinical presentations, which must be considered in our environment in case of progressive ataxia, ocular telangiectasias associated with an elevation of the serum alpha-fetoprotein level. Although there is currently no curative treatment, symptomatic and supportive management should be offered to these patients with the aim of improving the quality and expectancy of life. As with any chronic condition, educating patients and their families allows better adherence to treatment. As the first multicenter study on Ataxia-Telangiectasia carried out in Cameroon, it could pave the way for the development of a national registry that would allow better information regarding the disease for caregivers of children and lead to a more exhaustive collection of cases. | | | | Authors Contribution | | DE, CANK and SN designed the study, drafted the initial manuscript, reviewed and revised the manuscript. JMK, DNN, FKL and PCM designed the data collection instruments, collected the data, reviewed and revised the manuscript. DCK, EM and SN designed the study, coordinated, supervised data collection and critically reviewed the manuscript for important intellectual content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work. | | | | Compliance with Ethical Standards | | Funding | | This research did not receive a specific grant. | | | | Conflict of Interest | The authors declare that they have no competing interest.
Compliance with ethical standards
This study was carried out following the ethical principles of the Helsinki declaration. | | |
- Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM. Ataxia telangiectasia: a review. Orphanet J Rare Dis [Internet]. 2016 Nov 25 [cited 2020 Jul 31];11. Available from: https://www.ncbi.nlm.nih.gov/ pmc/articles/PMC5123280/ [CrossRef] [PubMed] [PMC free article]
- Chaudhary MW, Al-Baradie RS. Ataxia-telangiectasia: future prospects. Appl Clin Genet. 2014;7:159-167. [CrossRef] [PubMed] [PMC free article]
- Gatti RA, Berkel I, Boder E, Braedt G, Charmley P, Concannon P, et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22-23. Nature. 1988;336:577-580. [CrossRef] [PubMed]
- Swift M, Morrell D, Cromartie E, Chamberlin AR, Skolnick MH, Bishop DT. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am J Hum Genet. 1986;39:573-583.
- Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759-769. [CrossRef] [PubMed]
- Amirifar P, Ranjouri MR, Yazdani R, Abolhassani H, Aghamohammadi A. Ataxia-telangiectasia: A review of clinical features and molecular pathology. Pediatr Allergy Immunol [Internet]. 2019 [cited 2020 Aug 1];30:277-288. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/pai.13020 [CrossRef] [PubMed]
- Taylor AMR, Byrd PJ. Molecular pathology of ataxia telangiectasia. J Clin Pathol. 2005;58:1009-1015. [CrossRef] [PubMed] [PMC free article]
- Chun HH, Sun X, Nahas SA, Teraoka S, Lai CH, Concannon P, et al. Improved diagnostic testing for ataxia-telangiectasia by immunoblotting of nuclear lysates for ATM protein expression. Mol Genet Metab. 2003;80:437-443. [CrossRef] [PubMed]
- Hoche F, Seidel K, Theis M, Vlaho S, Schubert R, Zielen S, et al. Neurodegeneration in ataxia telangiectasia: what is new? What is evident? Neuropediatrics. 2012;43:119-129. [CrossRef] [PubMed]
- Alyasin S, Esmaeilzadeh H, Ebrahimi N, Nabavizadeh SH, Nemati H. Clinical Presentation of Ataxia-Telangiectasia. Arch Iran Med. 2019;22:682-686.
- Moin M, Aghamohammadi A, Kouhi A, Tavassoli S, Rezaei N, Ghaffari S-R, et al. Ataxia-telangiectasia in Iran: clinical and laboratory features of 104 patients. Pediatr Neurol. 2007;37:21-28. [CrossRef] [PubMed]
- Ayache R, Najjar MF, Obeid H, Pousse H, Gueddiche MN. Ataxia telangiectasia. Clinical and biological study in 17 cases. Ann Biol Clin (Paris). 1994;52:117-120.
- E. Mbonda, L. Dongmo, A. G. Juimo, H. Sile-Mefo, H. Nkoulou, E. Tetanye, et al. [ATAXIA-TELANGIECTASIA: A RARE AFFECTION OBSERVED IN A 6 YEAR OLD GIRL AND A 5 YEAR OLD BOY IN YAOUNDE]. Médecine d'Afrique Noire [Internet]. 1992;7:526-8.
- Pashankar F, Singhal V, Akabogu I, Gatti RA, Goldman FD. Intact T cell responses in ataxia telangiectasia. Clin Immunol Orlando Fla. 2006;120:156-162. [CrossRef] [PubMed]
- Regueiro JR, Porras O, Lavin M, Gatti RA. ATAXIA-TELANGIECTASIA: A Primary Immunodeficiency Revisited. Immunol Allergy Clin [Internet]. 2000 Feb 1 [cited 2020 Sep 11];20(1):177-206. Available from: https://www.immunology.theclinics.com/article/S0889-8561(05)70141-7/abstract [CrossRef]
- Nowak-Wegrzyn A, Crawford TO, Winkelstein JA, Carson KA, Lederman HM. Immunodeficiency and infections in ataxia-telangiectasia. J Pediatr. 2004;144:505-511. [CrossRef] [PubMed]
- Lefton-Greif MA, Crawford TO, Winkelstein JA, Loughlin GM, Koerner CB, Zahurak M, et al. Oropharyngeal dysphagia and aspiration in patients with ataxia-telangiectasia. J Pediatr. 2000;136:225-231. [CrossRef]
- Krauthammer A, Lahad A, Sarouk Y, Somech R, Nissenkorn A, Modan-Moses D, et al. Long-term nutritional and gastrointestinal aspects in patients with ataxia telangiectasia. Nutr Burbank Los Angel Cty Calif. 2018;46:48-52. [CrossRef] [PubMed]
- Woods CG, Taylor AM. Ataxia telangiectasia in the British Isles: the clinical and laboratory features of 70 affected individuals. Q J Med. 1992;82:169-179.
- Akturk H, Sutcu M, Somer A, Piskin S, Acar M, Ozmen M, et al. Ataxia telangiectasia in Turkey: multisystem involvement of 91 patients. World J Pediatr. 2017;13:465-471. [CrossRef] [PubMed]
- Stray-Pedersen A, Borresen-Dale AL, Paus E, Lindman CR, Burgers T, Abrahamsen TG. Alpha fetoprotein is increasing with age in ataxia-telangiectasia. Eur J Paediatr Neurol. 2007;11:375-380. [CrossRef] [PubMed]
- Tavani F, Zimmerman RA, Berry GT, Sullivan K, Gatti R, Bingham P. Ataxia-telangiectasia: the pattern of cerebellar atrophy on MRI. Neuroradiology. 2003;45:315-319. [CrossRef] [PubMed]
- Sahama I, Sinclair K, Pannek K, Lavin M, Rose S. Radiological imaging in ataxia telangiectasia: a review. Cerebellum Lond Engl. 2014;13:521-530. [CrossRef] [PubMed]
DOI: https://doi.org/10.7199/ped.oncall.2021.25
|
| Cite this article as: | | Enyama D, Ngo Kana C A, Mayouego Kouam J, Noukeu Njinkui D, Kemta Lekpa F, Mbonda P C, Kedy D C, Mbonda E, Nguefack S. Ataxia-Telangiectasia in Cameroonian Children. Pediatr Oncall J. 2021;18: 45-49. doi: 10.7199/ped.oncall.2021.25 |
|