Wing In Yam, Shirley Man Yee Wong, Betty Wai Man But.
Department of Paediatrics, Queen Elizabeth Hospital, Hong Kong SAR.
ADDRESS FOR CORRESPONDENCE
Dr. Wing In YAM, Department of Paediatrics, Queen Elizabeth Hospital, 30 Gascoigne Road, Kowloon, Hong Kong SAR.
Email: ivyivywingin@gmail.com | | Abstract | | Osteogenesis imperfecta Type XV, first reported in 2013, is caused by WNT1 gene mutations and is characterized by moderate to severe bone fragility and deformities with osteoporosis. We reported three cases of patients with WNT1-related osteogenesis imperfecta, who presented in early infancy and subsequently developed multiple fractures and bone deformities but with different extra-skeletal manifestations, including one patient with neonatal supraventricular tachycardia with Wolf-Parkinson-White syndrome, which was not reported in previous literature. | | | | Keywords | | WNT1, Osteogenesis imperfecta, extra-skeletal manifestations, OI Type XV. | | | | Introduction | | Osteogenesis imperfecta (OI) is an inherited skeletal dysplasia characterized by bone fragility and deformities. The majority of cases are caused by pathogenic variants encoding collagen Type 1 but alterations in genes not encoding collagen Type 1 account for 15-25% of cases.1 According to the Online Mendelian Inheritance in Man (OMIM) database, there are up to twenty-two types of osteogenesis imperfecta. OI Type XV is caused by mutations of the WNT1 gene, which is not related to collagen Type 1 synthesis but related to osteoblasts’ function. Here we report three patients with OI Type XV in recent years who had significant skeletal deformities and osteoporosis but with different extra-skeletal manifestations. | | | | Case Report | Patient 1
Our first patient is a 29-year-old Chinese lady. She presented at one month of age with a large anterior fontanelle, craniotabes, blue sclera and generalized hypotonia. The skeletal survey showed multiple long bone fractures including the left humerus, left clavicle and left-sided ribs with generalized osteopenia. She subsequently developed repeated fractures of the long bones with severe skeletal deformities and scoliosis (Figure 1a, 1b). She was wheelchair-dependent and her final height was 82 cm. She had mild ptosis. She developed severe restrictive lung disease with obstructive sleep apnoea requiring nocturnal bi-level positive airway pressure ventilation support. She also had right conductive hearing loss. Targeted next-generation sequencing (NGS) of OI gene panel was performed in 2015 when she was 22 and it identified compound heterozygous mutations of the WNT1(NM_005430.3) gene: c.506dupG, p.(Cys170Leufs*6), which had been reported as pathogenic in patients with OI type XV and a novel frameshift variant c.776_782dupGCAACCG, p.(Gly262Glnfs*57) at that time. She was put on regular pamidronate infusion when she was 13 years old for five years. Her bone pain and bone mineral density improved after pamidronate. However, she developed increased bone pain during the drug holiday and hence pamidronate was resumed (Graph 1). Despite pamidronate, she continued to develop new fractures.
Graph 1. Bone mineral density (BMD) of Patient 1 over L1-4 in relation to bisphosphonate treatment.
Solid line: z-score. Dotted line: bone mineral density in g/cm2.
.jpg)
Reference for z-score: Wu XP, Liao EY, Zhang H, Shan PF, Cao XZ, Liu SP. Establishment of BMD reference plots and determination of peak BMD at multiple skeletal regions in mainland Chinese women and the diagnosis of osteoporosis. Osteoporosis international. 2004 Jan;15(1):71-9.
Figure 1a. X-ray of Patient 1 with severe scoliosis when she was 15.
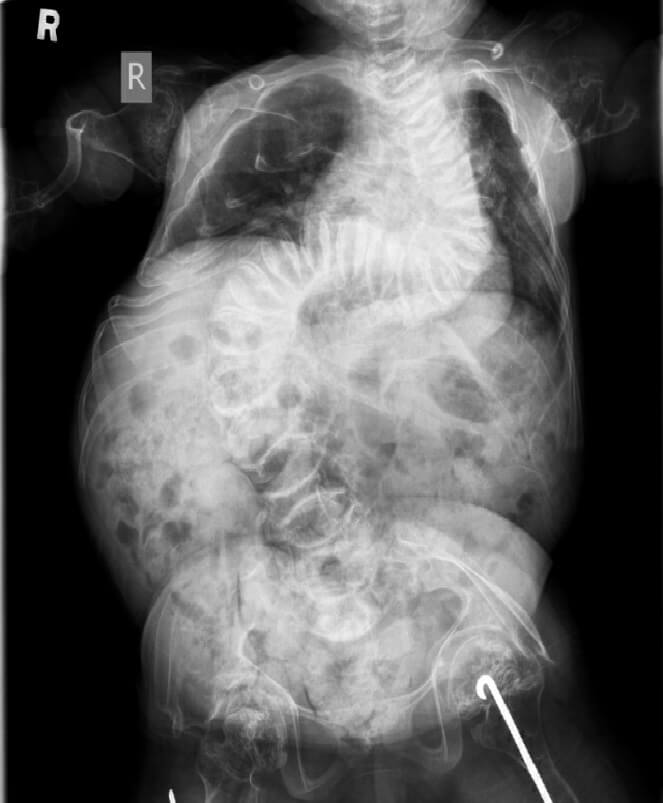
Figure 1b. X-ray of Patient 1 with lower limb deformities (arrows) when she was 26.
Patient 2
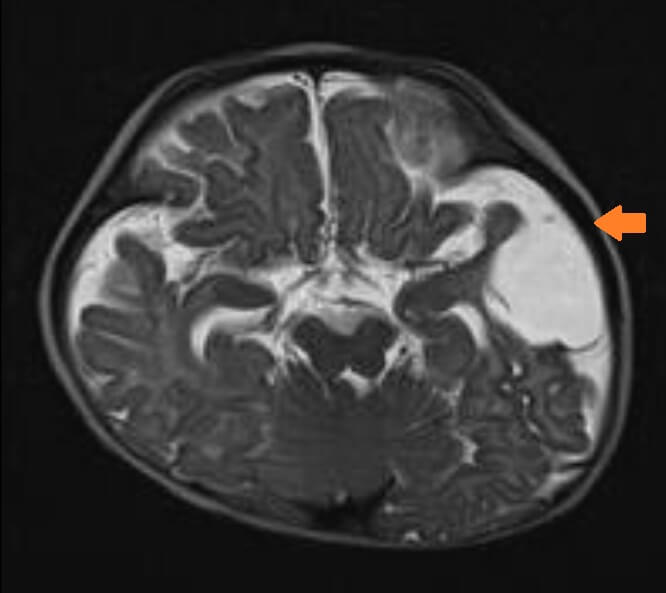
Our second patient was a 7-year-old Chinese boy. He was born at 35 weeks of gestation by spontaneous vaginal delivery with a birth weight of 3.13 kg. He had down-slanting palpebral fissures, ptosis and craniotabes. There was no blue sclera. He developed cardiac arrest on day 4 of life responsive to cardiopulmonary resuscitation for 3 minutes. He had Wolf-Parkinson-White (WPW) syndrome features on the electrocardiogram (Figure 2a). He developed an episode of supraventricular tachycardia at one month of life which was aborted by carotid massage and was subsequently put on propranolol. The echocardiogram did not review any structural abnormalities. He was first noted to have fractures of the left clavicle and left 2-4th ribs with osteopenia on the chest X-ray at one month of life. Within the first three months, he developed multiple fractures of the long bones without significant trauma. He was started on pamidronate infusion at 10 months of age. He continued to have fractures of the long bones 4-5 times per year documented by X-rays and significant osteoporosis. His family opted not for further pamidronate infusion when he was 4 years old because they did not find a significant effect of pamidronate on the patient and the central line for pamidronate infusion had been blocked. He suffered from significant bone deformity with bowing over the femurs, tibias and humerus with scoliosis (Figure 2b). Osteotomy was offered to the family. Whole exome sequencing was performed when he was three years old and noted compound heterozygous mutations of the WNT1(NM_005430.3) gene: heterozygous missense likely pathogenic variant c.385G>A, (p.Ala129Thr) and c.937C>T, (p.Arg313Cys), which was novel at the moment of analysis but subsequently reported to be related to osteogenesis imperfecta. Regarding the other systems, an MRI of the brain at four months of age showed widened basal cisterns with brachycephaly and a cystic lesion in the left temporal lobe compatible with a porencephalic cyst (Figure 2c), which could be due to previous insults. He suffered significant developmental delay, achieving one-year-old milestones when he was 6 years old. He was only able to sit on a specially-designed chair designed chair. He also had poorly formed incisors and canine with staining. The WPW syndrome features were no longer identified on the electrocardiogram after 2 months of age (Figure 2a) and there was no recurrence of the supraventricular tachycardia. propranolol was continued till the patient was 1.5 years of age. He recently developed snoring and was confirmed to have sleep-disordered breathing with desaturations on the overnight oximetry. He was put on nocturnal nasal continuous positive airway pressure ventilation.
Figure 2a. Electrocardiograms of Patient 2. The upper electrocardiogram showed the presence of delta waves suggestive of WPW syndrome. The PR interval and QRS duration were within the normal limit for age. The lower electrocardiogram was taken at 2 months of age, which did not show delta waves. An artifact was noted.
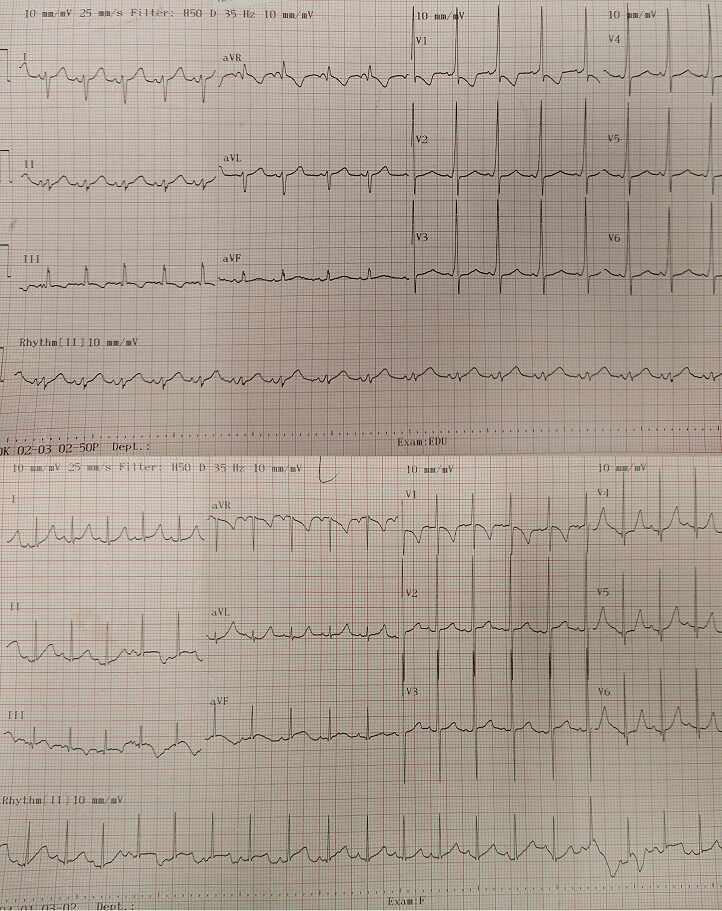
Figure 2b. X-ray of Patient 2 with significant lower limb deformities when he was 6.
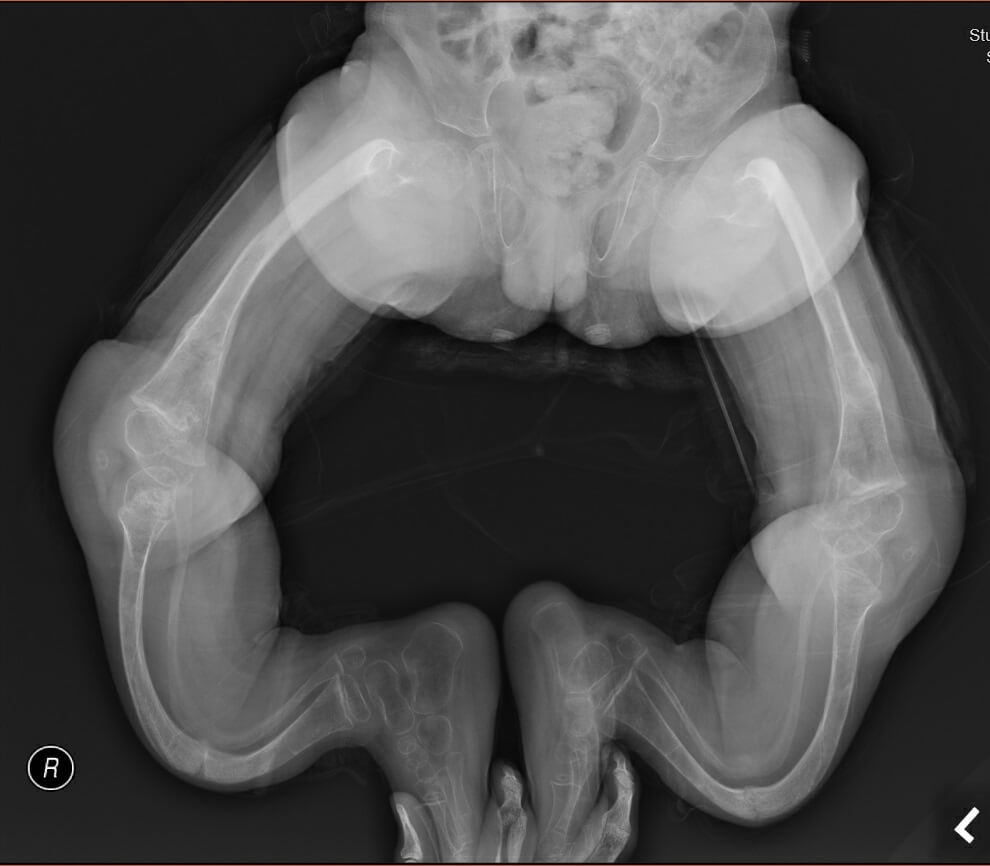
Figure 2c. T2 weighted MRI of Patient 2 at 4 months of age showed brachycephaly, widened basal cisterns and a left porencephalic cyst (shown by the arrow).
Patient 3
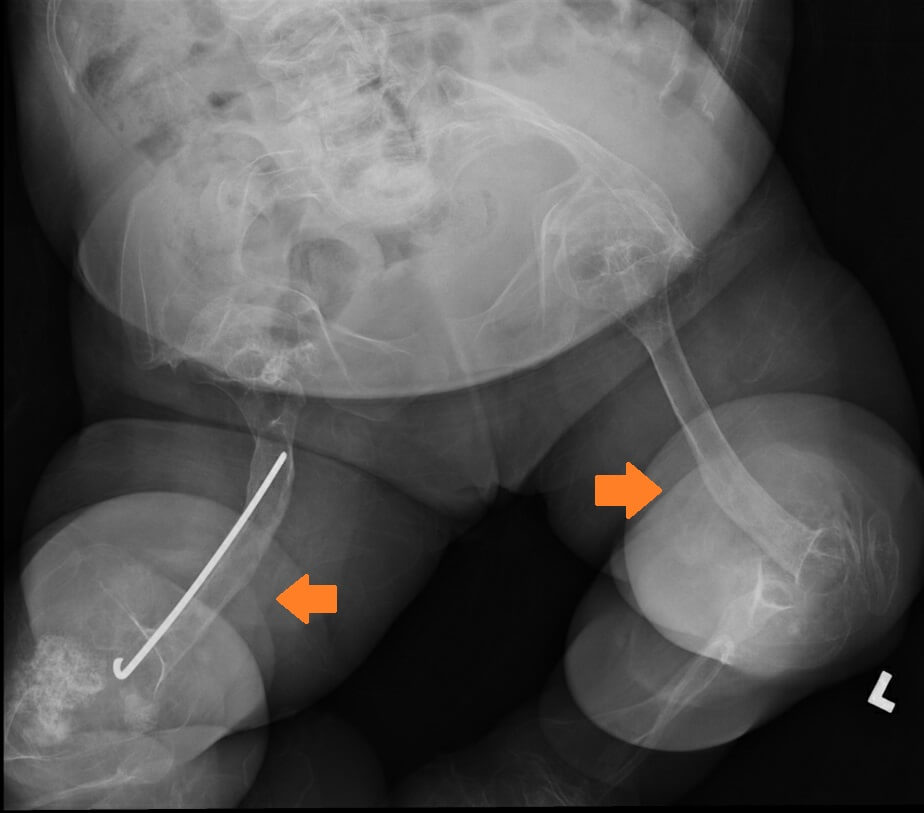
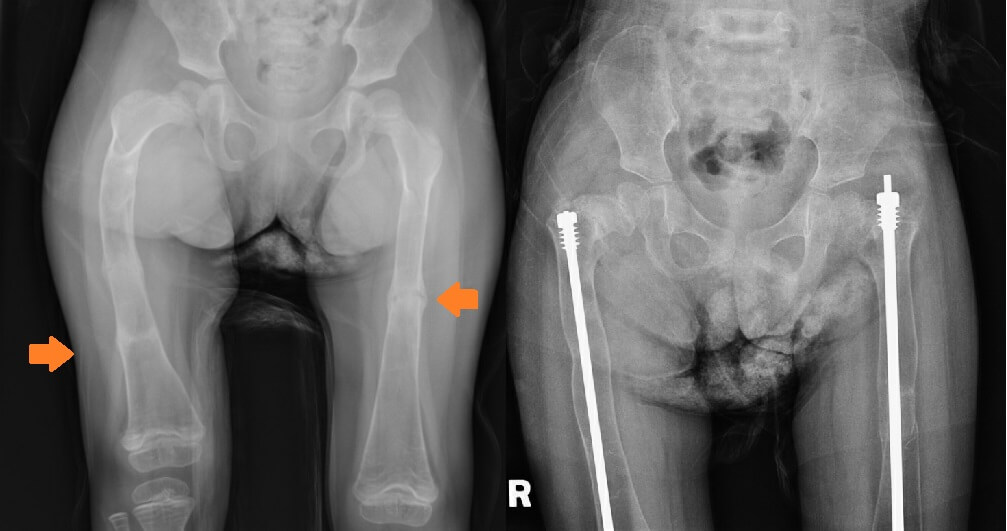
Our third patient is a 5-year-old Chinese girl who was born at full term by Caesarean Section with a fracture of the right clavicle. She presented at five months of age with a fractured mid-shaft of the left femur after a trivial injury. At 11 months of age, she fractured her right distal femur after falling from the bed. She did not have any blue sclera but she had bilateral ptosis. Targeted NGS revealed two heterozygous likely pathogenic variants in the WNT1 (NM_005430.3) gene: c.104+1G>A and c.216dupA, p.(Arg73Thrfs*82). Her mother was an unaffected carrier of the c.104+1G>A variant. She was then started on pamidronate infusion at 15 months of age. She continued to have fractures of the long bone four to five times per year and developed significant bone deformities (Figure 3) while the bone mineral density improved initially after pamidronate. She had limited mobility (standing with frame) but her development was otherwise normal. Her hearing was normal. The MRI of the brain was normal. Intramedullary rodding surgery of both lower limbs was performed at four years of age (Figure 3).
Figure 3. X-ray of the femurs before surgery showing bilateral lower limb curvatures and shortening of the right femur (Left, Fracture sites pointed by the arrows); and after surgery (Right) when Patient 3 was 5 years old.
| | | | Discussion | We reported three Hong Kong-Chinese patients with osteogenesis imperfecta Type XV. The clinical features were summarized in Table 1. All of them presented in early infancy and subsequently developed multiple fractures, osteoporosis and bone deformities resulting in limited mobility. However, their extra-skeletal manifestations were different which suggested that OI related to WNT1 mutations is a heterogeneous disease.
Osteogenesis imperfecta Type XV, first reported in 2013, is caused by mutations of the WNT1 gene located on chromosome 12q13. In mice studies, WNT1 is expressed during neuronal development and inactivation of it results in midbrain and cerebellar malformations in mice. WNT1 gene is also expressed in the osteoblast lineage and osteoblast activities decreased if there was a mutation in the WNT1 gene in mice models. In, in vitro studies, WNT1 protein binds to LRP5 (low-density lipoprotein receptor-related protein 5), which plays a role in the cell-fate decision, proliferation and differentiation of osteoblast lineage cells and activates the WNT-regulated β-catenin signaling cascade. Thus it is believed to be an important regulator of bone metabolism.2,3
Osteogenesis imperfecta Type XV is inherited in an autosomal recessive manner. It was characterized by early onset of multiple fractures during infancy or in utero, which subsequently resulted in bone deformities and significant osteoporosis. In our literature review of all journal articles available on PubMed related to WNT1 mutations-related osteogenesis imperfecta, 109 patients with clinical details were reported in 22 published journal articles from 2013 to 2022, including 45 male and 34 female patients (gender information was not available in 30 patients), age ranged from 0.8-32 years (median 6.35 years) at the time of publication.2,4-24 Most of them presented with fractures during early infancy (Range from in utero to 8 years, median 0.17 years) and had moderate to severe osteogenesis imperfecta with multiple fractures, which subsequently proceeded to bone deformities and osteoporosis.
Regarding the extra-skeletal manifestations, 39 patients had ptosis out of 61 cases (64%) with documented ptosis or not. Blue sclera was noted in 24 out of 95 cases (25%). 19 out of 91 (21%) had dentinogenesis imperfecta. Hearing impairment was uncommon with only 4 out of 72 cases (5.6%). 25% (16 out of 64) of them had intellectual disabilities or autism with two of them having seizures. Yet only 12 of them had brain imaging performed and 11 of them had abnormalities noted on the scans. One patient had cerebral atrophy but normal intelligence and one patient had autism but with a normal MRI. In fact, only 22 patients had their status of presence or absence of structural brain abnormalities reported in the studies and 12 of them (55%) had abnormal brain imaging findings. Among the 12 patients, five patients had cerebral atrophy only. Five patients had malformations or hypoplasia of the brainstem or cerebellum. One patient had an arachnoid cyst. One patient had frontal lobes atrophy and hypoplasia of the left brainstem and cerebellum and also the presence of an arachnoid cyst. None of the patients had cerebellar ataxia clinically. No association was noted between the genotype and phenotype with regard to neurological manifestations. Even patients in the same family could have different manifestations from normal intelligence to severe intellectual disability and brain malformations. The porencephalic cyst in Patient 2 was not reported in previous literature. However, it is not possible to differentiate whether it is congenital or acquired due to possible brain injuries during his neonatal period.
None of the reported cases had cardiac involvements. Patient 2 also had Wolf-Parkinson-White syndrome and neonatal supraventricular tachycardia. OI patients are prone to develop valvulopathies including mitral and aortic regurgitations, heart failure, atrial arrhythmia and aortic dilatations.25,26 Type 1 collagen is present in the heart valves, chordae tendineae, annuli fibrosis and the interventricular septum thus those with COL1A1/2 mutations are at risk of various heart conditions. However, previous studies did not mention the genotypes of OI patients hence it is not known whether the same association occurs in patients without collagen mutations. Two of our patients have sleep-disordered breathing which was not addressed in previous case reports on OI Type XV. Yet previous studies suggested that it is common in patients with severe OI with bone deformities.27
Intravenous bisphosphonates are recommended for use in children with severe osteogenesis imperfecta. Cochrane review showed a universal improvement in bone density. However, data on fracture reduction, bone pain, growth and functional improvement were incomplete.28 The effect of bisphosphonates on reduction in fractures or bone deformities was not substantial in previously reported OI type XV cases but improvement in bone density had been reported.2,4,5,6,7,8 In contrast to COL1A1/2 mutations which increase bone turnover, WNT1 mutations affect osteoblasts’ functions and reduce bone formation. Bisphosphonate only blocks the action of osteoclasts and decreases bone resorption. It has no effects on osteoblasts’ functions. Sclerostin is a protein secreted by osteocytes that blocks the WNT1 signaling and has anti-anabolic effects on the bone.29 The use of sclerostin inhibitors in the treatment of osteoporosis has already been approved. It is currently under clinical trials for the treatment of OI in adult patients.1
Table 1. Comparison of the phenotypes of the three patients with reported cases in the literature.
| Clinical features |
Patient 1 |
Patient 2 |
Patient 3 |
Reported cases* (Total=109) |
| First presentation |
1 month |
1 month |
5 months |
Early infancy (Median 0.17 years) |
| Current age (years) ^ |
29 |
5 |
0.8-32 |
| Multiple fractures |
Yes |
Yes |
Yes |
Yes |
| Osteoporosis |
Yes |
Yes |
Yes |
Yes |
| Bone deformities |
Yes |
Yes |
Yes |
Yes |
| Mobility |
Limited |
Limited |
Limited |
Usually limited |
| Response to bisphosphonates |
Improved bone pain and bone mineral density |
No significant response |
Initial response in bone mineral density |
Some responses in bone mineral density |
| Extra-skeletal manifestations |
| Clinical features |
Patient 1 |
Patient 2 |
Patient 3 |
Reported cases |
| Blue sclera |
Yes |
No |
No |
25% (24/95) |
| Ptosis |
Yes |
Yes |
Yes |
64% (39/61) |
| Hearing impairment |
Yes |
Not noted |
Not noted |
5.6% (4/72) |
| Dentinogenesis imperfecta |
No |
Yes |
No |
21% (19/91) |
| Intellectual disability |
No |
Yes |
No |
25% (16/64) |
| Structural brain abnormalities |
No |
Widened basal cistern with a cystic lesion in the left temporal lobe |
No |
Any abnormality: 55% (12/22)
5 patients with cerebral atrophy; 5 patients with brainstem/cerebellar malformations; 1 patient with an arachnoid cyst; 1 patient with cerebral atrophy with brainstem and cerebellar malformations and an arachnoid cyst |
| Sleep-disordered breathing |
Yes, on BiPAP |
Yes, on CPAP |
No |
Not reported |
| Cardiac involvement |
No |
WPW syndrome |
No |
Cardiac complications not reported |
*Ratios denote the number of patients showing a particular phenotype / total number of patients with that phenotype described
^Age at the time of manuscript preparation
| | | | Conclusion | | We reported three patients with osteogenesis imperfecta type XV. All of them presented in infancy with multiple fractures, severe bone deformities and osteoporosis. They had variable extra-skeletal manifestations. One of them was intellectually disabled and he also had Wolf-Parkinson-White syndrome, which was not reported in previous literature. pamidronate did not seem to prevent fractures in our patients. Novel therapies targeting osteoblasts’ functions hopefully will bring hope to these patients in the future. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Marom R, Rabenhorst BM, Morello R. Osteogenesis imperfecta: an update on clinical features and therapies. European Journal of Endocrinology. 2020 Oct 1;183(4):R95-106. [CrossRef] [PubMed] [PMC free article]
- Keupp K, Beleggia F, Kayserili H, Barnes AM, Steiner M, Semler O, Fischer B, Yigit G, Janda CY, Becker J, Breer S. Mutations in WNT1 cause different forms of bone fragility. The American Journal of Human Genetics. 2013 Apr 4;92(4):565-74. [CrossRef] [PubMed] [PMC free article]
- Joeng KS, Lee YC, Jiang MM, Bertin TK, Chen Y, Abraham AM, Ding H, Bi X, Ambrose CG, Lee BH. The swaying mouse as a model of osteogenesis imperfecta caused by WNT1 mutations. Human molecular genetics. 2014 Aug 1;23(15):4035-42. [CrossRef] [PubMed] [PMC free article]
- Aldinger KA, Mendelsohn NJ, Chung BH, Zhang W, Cohn DH, Fernandez B, Alkuraya FS, Dobyns WB, Curry CJ. Variable brain phenotype primarily affects the brainstem and cerebellum in patients with osteogenesis imperfecta caused by recessive WNT1 mutations. Journal of medical genetics. 2016 Jun 1;53(6):427-30. [CrossRef] [PubMed] [PMC free article]
- Pyott SM, Tran TT, Leistritz DF, Pepin MG, Mendelsohn NJ, Temme RT, Fernandez BA, Elsayed SM, Elsobky E, Verma I, Nair S. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. The American Journal of Human Genetics. 2013 Apr 4;92(4):590-7. [CrossRef] [PubMed] [PMC free article]
- Fahiminiya S, Majewski J, Mort J, Moffatt P, Glorieux FH, Rauch F. Mutations in WNT1 are a cause of osteogenesis imperfecta. Journal of medical genetics. 2013 May 1;50(5):345-8. [CrossRef] [PubMed]
- Nampoothiri S, Guillemyn B, Elcioglu N, Jagadeesh S, Yesodharan D, Suresh B, Turan S, Symoens S, Malfait F. Ptosis as a unique hallmark for autosomal recessive WNT1‐associated osteogenesis imperfecta. American Journal of Medical Genetics Part A. 2019 Jun;179(6):908-14. [CrossRef] [PubMed]
- Liu Y, Song L, Ma D, Lv F, Xu X, Wang J, Xia W, Jiang Y, Wang O, Song Y, Xing X. Genotype-phenotype analysis of a rare type of osteogenesis imperfecta in four Chinese families with WNT1 mutations. Clinica Chimica Acta. 2016 Oct 1;461:172-80. [CrossRef] [PubMed]
- Laine CM, Joeng KS, Campeau PM, Kiviranta R, Tarkkonen K, Grover M, Lu JT, Pekkinen M, Wessman M, Heino TJ, Nieminen-Pihala V. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. New England Journal of Medicine. 2013 May 9;368(19):1809-16. [CrossRef] [PubMed] [PMC free article]
- Palomo T, Al-Jallad H, Moffatt P, Glorieux FH, Lentle B, Roschger P, Klaushofer K, Rauch F. Skeletal characteristics associated with homozygous and heterozygous WNT1 mutations. Bone. 2014 Oct 1;67:63-70. [CrossRef] [PubMed]
- Panigrahi I, Didel S, Kirpal H, Bellampalli R, Miyanath S, Mullapudi N, Rao S. Novel mutation in a family with WNT1-related osteoporosis. European Journal of Medical Genetics. 2018 Jul 1;61(7):369-71. [CrossRef] [PubMed]
- Lu Y, Dai Y, Wang Y, Zhai N, Zhang J, Liu J, Yin X, Li T, Ren X, Han J. Complex heterozygous WNT1 mutation in severe recessive osteogenesis imperfecta of a Chinese patient. Intractable & Rare Diseases Research. 2018 Feb 28;7(1):19-24. [CrossRef] [PubMed] [PMC free article]
- Chen P, Chen J, Yang Z, Lu Y, Shen L, Zhou K, Ye S, Shen B. Consanguineous‐derived homozygous WNT1 mutation results in osteogenesis imperfect with congenital ptosis and exotropia. Molecular Genetics & Genomic Medicine. 2020 Aug;8(8):e1350. [CrossRef] [PubMed] [PMC free article]
- Kausar M, Siddiqi S, Yaqoob M, Mansoor S, Makitie O, Mir A, Khor CC, Foo JN, Anees M. Novel mutation G324C in WNT1 mapped in a large Pakistani family with severe recessively inherited Osteogenesis Imperfecta. Journal of biomedical science. 2018 Dec;25(1):1-0. [CrossRef] [PubMed] [PMC free article]
- Faqeih E, Shaheen R, Alkuraya FS. WNT1 mutation with recessive osteogenesis imperfecta and profound neurological phenotype. Journal of medical genetics. 2013 Jul 1;50(7):491-2. [CrossRef] [PubMed]
- Lu Y, Ren X, Wang Y, Bardai G, Sturm M, Dai Y, Riess O, Zhang Y, Li H, Li T, Zhai N. Novel WNT1 mutations in children with osteogenesis imperfecta: Clinical and functional characterization. Bone. 2018 Sep 1;114:144-9. [CrossRef] [PubMed]
- Kantaputra PN, Sirirungruangsarn Y, Visrutaratna P, Petcharunpaisan S, Carlson BM, Intachai W, Sudasna J, Kampuansai J, Dejkhamron P. WNT1-associated osteogenesis imperfecta with atrophic frontal lobes and arachnoid cysts. Journal of Human Genetics. 2019 Apr;64(4):291-6. [CrossRef] [PubMed]
- Kuptanon C, Srichomthong C, Sangsin A, Kovitvanitcha D, Suphapeetiporn K, Shotelersuk V. The most 5′ truncating homozygous mutation of WNT1 in siblings with osteogenesis imperfecta with a variable degree of brain anomalies: a case report. BMC Medical Genetics. 2018 Dec;19(1):1-5. [CrossRef] [PubMed] [PMC free article]
- Li S, Cao Y, Wang H, Li L, Ren X, Mi H, Wang Y, Guan Y, Zhao F, Mao B, Yang T. Genotypic and phenotypic analysis in Chinese cohort with autosomal recessive osteogenesis imperfecta. Frontiers in genetics. 2020:984. [CrossRef] [PubMed] [PMC free article]
- Won JY, Jang WY, Lee HR, Park SY, Kim WY, Park JH, Kim Y, Cho TJ. Novel missense loss-of-function mutations of WNT1 in an autosomal recessive osteogenesis imperfecta patient. European journal of medical genetics. 2017 Aug 1;60(8):411-5. [CrossRef] [PubMed]
- Stephen J, Girisha KM, Dalal A, Shukla A, Shah H, Srivastava P, Kornak U, Phadke SR. Mutations in patients with osteogenesis imperfecta from consanguineous Indian families. European journal of medical genetics. 2015 Jan 1;58(1):21-7. [CrossRef] [PubMed]
- Mrosk J, Bhavani GS, Shah H, Hecht J, Krüger U, Shukla A, Kornak U, Girisha KM. Diagnostic strategies and genotype-phenotype correlation in a large Indian cohort of osteogenesis imperfecta. Bone. 2018 May 1;110:368-77. [CrossRef] [PubMed]
- Hayat A, Hussain S, Bilal M, Kausar M, Almuzzaini B, Abbas S, Tanveer A, Khan A, Siddiqi S, Foo JN, Ahmad F. Biallelic variants in four genes underlying recessive osteogenesis imperfecta. European journal of medical genetics. 2020 Aug 1;63(8):103954. [CrossRef] [PubMed]
- Umair M, Alhaddad B, Rafique A, Jan A, Haack TB, Graf E, Ullah A, Ahmad F, Strom TM, Meitinger T, Ahmad W. Exome sequencing reveals a novel homozygous splice site variant in the WNT1 gene underlying osteogenesis imperfecta type 3. Pediatric research. 2017 Nov;82(5):753-8. [CrossRef] [PubMed]
- Ashournia H, Johansen FT, Folkestad L, Diederichsen AC, Brixen K. Heart disease in patients with osteogenesis imperfecta-a systematic review. International journal of cardiology. 2015 Oct 1;196:149-57. [CrossRef] [PubMed]
- Folkestad L, Hald JD, Gram J, Langdahl BL, Hermann AP, Diederichsen AC, Abrahamsen B, Brixen K. Cardiovascular disease in patients with osteogenesis imperfecta-a nationwide, register-based cohort study. International journal of cardiology. 2016 Dec 15;225:250-7. [CrossRef] [PubMed]
- Léotard A, Taytard J, Aouate M, Boule M, Forin V, Lallemant-Dudek P. Diagnosis, follow-up and management of sleep-disordered breathing in children with osteogenesis imperfecta. Annals of physical and rehabilitation medicine. 2018 May 1;61(3):135-9. [CrossRef] [PubMed]
- Simm PJ, Biggin A, Zacharin MR, Rodda CP, Tham E, Siafarikas A, Jefferies C, Hofman PL, Jensen DE, Woodhead H, Brown J. Consensus guidelines on the use of bisphosphonate therapy in children and adolescents. Journal of paediatrics and child health. 2018 Mar 1;54(3):223-33. [CrossRef] [PubMed]
- Lewiecki EM. Role of sclerostin in bone and cartilage and its potential as a therapeutic target in bone diseases. Therapeutic advances in musculoskeletal disease. 2014 Apr;6(2):48-57. [CrossRef] [PubMed] [PMC free article]
DOI: https://doi.org/10.7199/ped.oncall.2024.17
|
| Cite this article as: | | Yam W I, Wong S M Y, But B W M. Three Chinese Patients with WNT1 Mutations-related Osteogenesis Imperfecta and Literature Review of Extra-skeletal Manifestations. Pediatr Oncall J. 2024;21: 68-74. doi: 10.7199/ped.oncall.2024.17 |
|