Ramya Madhavan, Senthilkumar Palaniappan.
Masonic medical centre for children, Coimbatore, India.
ADDRESS FOR CORRESPONDENCE
Ramya Madhavan, Masonic medical centre for children Coimbatore-641018.
Email: drramyavijay82@gmail.com. | | Abstract | Alkaptonuria is a rare inherited condition of tyrosine metabolism. It is characterised by blackening of urine on contact with air. We herein report a case of nephrotic syndrome in a child with alkaptonuria. A 10 years old female child who was diagnosed as nephrotic syndrome at 2 years of age presented with anasarca, loose stools and decreased urine output. Investigations were suggestive of relapse of nephrotic syndrome. There was also history of urine colour turning black on standing since infancy and further investigations were done for blackening of urine. Urine Benedict’s test was positive and clinical exome sequencing was suggestive of alkaptonuria. The objective of this case report is to highlight usefulness of Benedict test’s as a screening test in suspected alkaptonuria, in situations where other screening tests and urine chromatography are not available. It is also to report the rare occurrence of nephrotic syndrome in a child with alkaptonuria which is not yet reported in literature.
| | | | Keywords | | Nephrotic syndrome, Alkaptonuria, blackening of urine. | | | | Case Report | This 10 years old female child was admitted with swelling around eyes, swelling of both feet with abdominal distension for 1 week and loose stools with decreased urine output for the previous 2 days. She is a known case of nephrotic syndrome diagnosed at the age of 2 years and so far relapsed once at six months after the first episode. Clinical examination revealed an alert and active child with anasarca. All growth parameters were within normal range for age. Her vitals were stable with heart rate: 100/min, respiratory rate: 30/min, BP: 106/80 mmHg, oxygen saturation: 99% in room air. Systemic examination revealed free fluid in the abdomen and other systems were normal. Urine routine showed 4+ proteinuria with urine spot PCR of 55:1. Complete blood count was normal, serum albumin was 1.2 gm%, blood urea was 19 mg%, serum creatinine was 0.4 mg%. She was diagnosed as minimal change nephrotic syndrome- second relapse.
Mother also gave a history of diaper turning into black after voiding since infancy. Alkaptonuria was suspected and Benedict’s test was done as a screening test. It was positive and Urine dipstrip for glucose was negative. Since urine chromatographic estimation of homogentisic acid(HGA) was not available, exome sequencing was done which showed homozygous mutation in homogentisate 1,2 – dioxygenase(HGD) gene confirming the diagnosis of alkaptonuria.
Figure 1. Clinical exome sequencing.
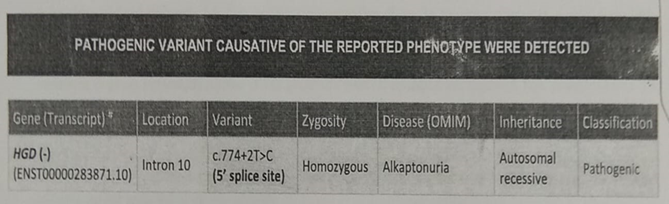
Figure 2. Positive Benedict's test.

Figure 3. Dark urine on standing (first container) and urine immediately after collection (second container).
 | | | | Discussion | Alkaptonuria is a rare inherited disorder of tyrosine metabolism. It was one of the first disorders known to transmit in a typical Mendelian recessive inheritance.1 It is due to deficiency of enzyme homogentisate 1, 2-dioxigenase and gene for the enzyme maps to chromosome 3q13.3.2
Deficiency of homogentisate 1,2-dioxigenase causes accumulation of homogentisic acid which inturn is excreted in urine or deposited in tissues.2 Hence, important clinical features are ochronosis ,ochronotic arthropathy and homogentisic aciduria.1
Ochronosis is accumulation of the black polymer of homogentisic acid which is seen clinically as dark spots on sclera and ear cartilage.2 This pigmentation of the sclera or the cartilage of the ear usually starts around 3rd decade and becomes prominent by 4th decade. Ochronotic arthropathy starts around 4th decade with involvement of spine, knees and shoulder joints resulting in narrowing of joint spaces.3 It resembles ankylosing spondylitis but it can be differentiated from ankylosing spondylitis by sparing of sacroiliac joint and also its by its radiographic appearance.4
Grey or black staining of the nappy and blackening of urine on standing are important clues during childhood as in this child.2 This darkening of urine is caused by oxidation of homogentisic acid to benzoquinoneacetate which forms a melanin-like polymer that slowly turns urine black.1 HGA being a reducing substance gives positive results with Benedict’s reagent, strong alkali and ferric chloride. Hence Benedict’s test, ferric chloride test and ammoniac silver nitrate tests are useful screening tests. HGA levels in the urine can be measured by gas chromatography-mass spectrometry or colorimetric acid analysis which confirms the diagnosis.5 Molecular diagnosis is based on detection of pathogenic variants.4
Adults with alkaptonuria may develop complications involving multiple organ systems including calcification of heart valves, renal stones formation, prostate stones formation and hypothyroidism.4
There is no definitive treatment for alkaptonuria. Surveillance for complications should begin by 4th decade.3 Palliative therapy includes physiotherapy, joint replacement surgery and pain control.1 A triketone herbicide, nitisinone inhibits 4-hydroxyphenylpyruvate dioxygenase, the enzyme that produces HGA but nitisinone increases tyrosine levels.3 The other option is vitamin C (Ascorbic acid) .Being an antioxidant, it inhibits oxidation of HGA but, it does not reduce urinary HGA excretion. Moreover ascorbic acid increases HGA production and contributes to the formation of renal oxalate stones.1 In presymptomatic children, treatment with nitisinone along with phenylalanine and tyrosine restricted diet is a reasonable option although no experience is available regarding long term efficacy and safety.2
| | | | Conclusion | | There are few other disorders reported in literature found to be associated with alkaptonuria including beta thalassemia, renal tubular acidosis, Pompes disease and Gilbert syndrome.6,7,8,9 Here, minimal change nephrotic syndrome is associated with alkaptonuria which to our knowledge is not yet reported in literature. This case is reported for this rare association and also to highlight the importance of Benedict test as a useful screening test in resource poor settings. Benedict’s test give positive result for reducing sugars. HGA being a reducing substance also gives positive result which can be counterchecked by negative urine dipstrip for glucose. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Mistry JB, Bukhari M & Taylor AM (2013) Alkaptonuria, Rare Diseases, 1:1, e27475, DOI: 10.4161/rdis.27475. [CrossRef] [PubMed] [PMC free article]
- Kliegman RM, St GemeJW, Blum NJ., et al. Nelson text book of pediatrics. 21st ed. Philadelphia:Elsevier; 2020.p.700-701.
- Tharini G, Ravindran V, Hema N, Prabhavathy D, Parveen B. Alkaptonuria. Indian J Dermatol. 2011 Mar;56(2):194-6. doi: 10.4103/0019-5154.80415. PMID: 21716546; PMCID: PMC3108520. [CrossRef] [PubMed] [PMC free article]
- Introne WJ, Gahl WA. Alkaptonuria. 2003 May 9 [Updated 2016 May 12]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2021. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1454/
- Krishan Nilantha Hewa Thalagahage,Jayaweera Arachchige Asela Sampath Jayaweera and Indika Senavirathne.2015.Detection of alkaptonuria in a 1 week old infant.BMJ case reports.DOI;10.1136/bcr-2014-208505. [CrossRef] [PubMed] [PMC free article]
- Lodh M, Kerketta JA. Early diagnosis of co-existent ß-thalassemia and alkaptonuria. Indian J Hum Genet. 2013;19(2):259-261. doi:10.4103/0971-6866.116104. [CrossRef] [PubMed] [PMC free article]
- Nickavar A, Azar MR. Alkaptonuria, a new association of distal renal tubular acidosis. Saudi J Kidney Dis Transpl. 2018 Jul-Aug;29(4):997-999. doi: 10.4103/1319-2442.239645. PMID: 30152443. [CrossRef] [PubMed]
- Habbal MZ, Bou Assi T, Mansour HAlkaptonuria and pompe disease in one patient: metabolic and molecular analysisCase Reports 2013;2013:bcr2012008491 [CrossRef] [PubMed] [PMC free article]
- Brown NK, Smuckler EA. Alkaptonuria and Gilbert's syndrome. Report of two affected siblings and hepatic ultrastructure in one sibling. Am J Med. 1970 Jun;48(6):759-65. doi: 10.1016/s0002-9343(70)80011-0. PMID: 5420562. [CrossRef] [PubMed]
DOI: https://doi.org/10.7199/ped.oncall.2025.18
|
| Cite this article as: | | Madhavan R, Palaniappan S. Nephrotic syndrome in a child with alkaptonuria. Pediatr Oncall J. 2025;22: 87-88. doi: 10.7199/ped.oncall.2025.18 |
|