Rajniti Prasad, O P Mishra, Narendra Reddy, Mahendra N Singh.
Department of Pediatrics, Institute of Medical Sciences, Banaras Hindu University, Varanasi, UP, India.
ADDRESS FOR CORRESPONDENCE
Dr.Rajniti Prasad, Lecturer, Department of Pediatrics, Institute of Medical Sciences, Banaras Hindu University, Varanasi, UP, India. PIN-221005
Email: rajnitip@yahoo.co.in | | Abstract | | Romano-ward syndrome is an autosomal dominant disorder characterized by prolonged QT interval, which may present with cardiac events such as syncope, cardiac arrest and sudden death in healthy children. We present this case who presented as cardiac asystole. | | | | Introduction | | Romano-ward syndrome is an autosomal dominant disorder characterized by prolonged QT interval, which may present with cardiac events such as syncope, cardiac arrest and sudden death in healthy children (1) . We present this case because of rarity of this condition and index child presenting as cardiac asystole, who had survived with cardiopulmonary resuscitation and is symptom-free on atenolol therapy. | | | | Case Report | 5 years male child presented with fever, cough, running nose for one day, vomiting (2 episodes) for 1 hour and altered sensorium with gasping respiration for 5 minutes. There is no history of drug intake, choking, convulsions and similar episodes in the past. There is no family history of deafness, sudden death, syncope or convulsions. On examination, child was cyanosed with occasional gasping respiration and heart sounds were not audible. Endotracheal intubation was done immediately and cardiopulmonary resuscitation was started. ECG leads were attached, which showed absence of electrical activity. Intravenous adrenaline was given (1:10000 dilution: 0.1ml/Kg) and cardiopulmonary resuscitation continued. Repeat dose of adrenaline was given one minute after 1st dose. After 2 minutes of 2nd dose of IV adrenaline, sinus rhythm with a heart rate of 130/minute was restored. Child became fully conscious in 30 minutes. ECG done showed prolonged QTc (0.61sec), inverted T waves in all the precordial leads, and a large amplitude inverted T wave, which was followed by a small amplitude T wave (V4, V5) or upright/flat T waves (L2) (fig-1).
Figure-1. ECG showing prolonged QT-interval with inverted T-wave
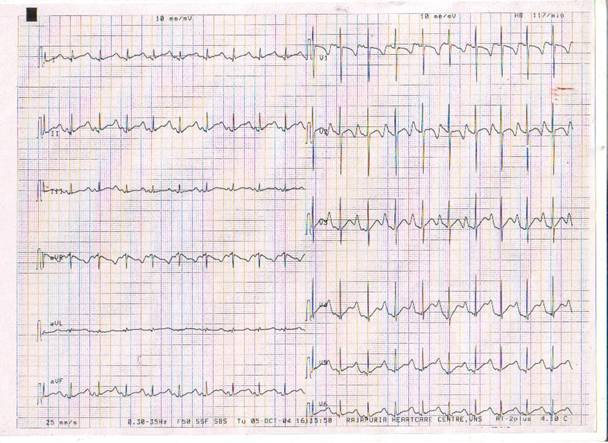
Figure-2. ECG repeated after 24 hrs showed prolonged QTc and large bizarre notched T waves
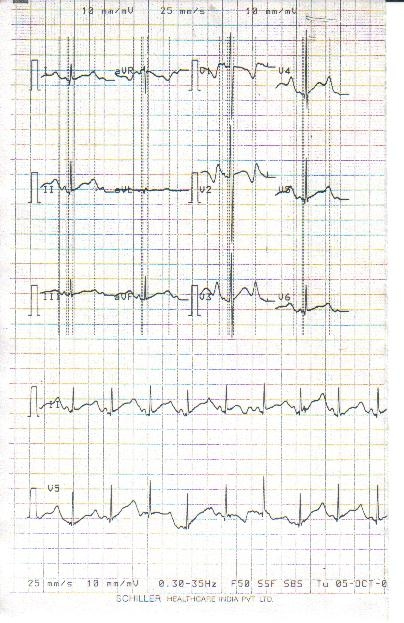
Serum calcium, phosphate, sodium, potassium and magnesium were normal. 24 hours Holter monitoring showed a minimum heart rate of 52/minutes, T wave changes mentioned above and no ventricular arrhythmia. Audiometric examination of the child was normal. ECG of the siblings and parents showed prolonged QTc (>0.47sec) in two sisters and mother (>0.52 sec). Mother is asymptomatic and had never experienced any problem. ECG of the grandparent could not be done. None of the parents and siblings had deafness on audiometric study. We have not done mutational study and DNA sequencing in either index case or family members as this facility is not available in our institute. The diagnosis of congenital Long QT syndrome (Romano-Ward syndrome) was made.
Since the child presented with a life-threatening event, attendants were given the choice of an Implantable Cardiac Defibrillator (ICD) Vs Beta blockers. As they could not afford an ICD, the child and his two sisters were put on atenolol. | | | | Discussion | Romano-Ward Syndrome is an autosomal dominant disorder characterized by syncopal and fainting attacks often precipitated by physical and emotional stress (fear, anger, loud noise, medications, sudden awakening) (2) but none were present in our patient. The onset of symptoms usually occurs in first two decades of life as in our index case but can be delayed especially in females. Electrocardiography shows prolonged repolarisation (long QT interval), abnormal morphology of T waves or a characteristic polymorphic ventricular tachycardia called torsades de pontes (3) . Polymorphic ventricular tachycardia is thought to be initiated by early after depolarization in purkinje system and maintained by re-entry in myocardium (2) . The severity of clinical manifestations may vary from a full blown disease with markedly prolonged QT interval and recurrent syncope to sub-clinical form with borderline QT prolongation (4) .
In pregenomic era, sympathetic imbalance was thought to be responsible for the disease. Since 1991 (postgenomic era) Romano-ward syndrome is caused by mutations of cardiac ion channel genes. Based on chromosomal loci and gene mutations, there are seven types of Romano-ward syndrome (2) .
| LQTS Type |
Chromosomal Locus |
Mutated gene |
Ion current affected |
| LQT1 |
11p15.5 |
KVLQT1 (KCNQ1) |
Potassium current(Iks) |
| LQT2 |
7q35-36 |
KCNH2 |
Potassium current(Iks) |
| LQT3 |
3p21-23 |
SCN5A |
Sodium current(I Na ) |
| LQT4 |
4q25-27 |
ANKB |
- |
| LQT5 |
21q21.1-22.2 |
KCNE1 |
Potassium current(Iks) |
| LQT6 |
21q22.1-22.2 |
KCNE2 |
Potassium current(Ikr |
| LQT7 |
17q23 |
KCNJ2 |
Potassium current(Kir 2.1) |
LQT1 and LQT2 are estimated to account for approximately 87% of cases with LQT3 accounts for 8%, whereas LQT5 and LQT6 are extremely rare. LQT7 is also known as Andersen syndrome (5) . It is a potassium sensitive periodic paralysis with low set ears, micrognathia and clinodactyly as well as long QT interval and ventricular arrhythmia. Despite advancement in molecular genetic knowledge, diagnosis is still based on electrocardiography and clinical characteristics. Beta-blockers remain the mainstay of treatment (6) . For high-risk patients, the implantable cardiac defibrillator (ICD) offers an effective therapeutic option to reduce mortality (7) . Gene based specific therapy is still preliminary (8) . | | | | Conclusion | | Patients with Romano-ward syndrome usually presents with sudden cardiac events in healthy children which may be prevented by ICD and /or beta-blockers. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Bloise R, Napolitano C, Priori SG. Romano-Ward and other congenital long QT syndromes. Cardiovasc Drugs Ther 2002; 16: 19-23. [CrossRef] [PubMed]
- Schwartz PJ, Priori SG, Spazolini C. Genotype-Phenotype correlation in long QT syndrome threatening arrhythmias. Circulation 2001; 103: 89-95. [CrossRef] [PubMed]
- Schwartz PJ, Priori SG, Napolitano C. The long QT syndrome cardiac electrophysiology : from cell to bedside. Philadelphia , WB Saunders, 2000 pp 597-615.
- Priori SG, Napolitano C, Schwartz PJ. Low penetrance in long QT syndrome: clinical impact. Circulation 1999; 99, 529. [CrossRef] [PubMed]
- Plaster NM, Jawel R, et al. Mutations in Kir-2.1 cause the developmental and episodic electrical phenotype of Andersen's syndrome. Cell 2001; 105: 511-19. [CrossRef]
- Widekind H, Schwarz M et al. Effective long term control of cardiac events with beta-blockers in a family with a common LQ1 mutation. Clin Genet 2004; 65: 233-41. [CrossRef]
- Viskin S, Fish R, Roth A et al. Influence of genotype on the clinical course of the long QT syndrome. Int. LQT syndrome Registry Research Group N Engl J Med 1998; 334: 960-965.
- Chiang CE. Congenital and acquired long QT syndrome. Current concepts and management. Cardiol Rev 2004; 12: 222-34. [CrossRef]
|
| Cite this article as: | | Prasad R, Mishra O P, Reddy N, Singh M N. Romano-Ward Syndrome in a Family with Index Case Presenting as Cardiac Asystole. Pediatr Oncall J. 2007;4: 17. |
|