Madalena Almeida Borges1, Maria Costa1, Rute Baeta Baptista1, Ana Laura Fitas2, Telma Francisco1, Margarida Abranches1.
1Pediatric Nephrology Unit, Hospital Dona Estefânia, Centro Hospitalar Universitário de Lisboa Central, Lisbon, Portugal,
2Pediatric Endocrinology Unit, Hospital Dona Estefânia, Centro Hospitalar Universitário de Lisboa Central, Lisbon, Portugal.
ADDRESS FOR CORRESPONDENCE
Madalena Almeida Borges, Rua Jacinta Marto, 8A, 1169-045 Lisboa, Portugal.
Email: madalenalmeidaborges@gmail.com | | Abstract | X-linked hypophosphatemia (XLH) is the most common form of genetic rickets and it is caused by mutations in the phosphate regulating endopeptidase homolog X-linked (PHEX).
We present a case of a five-year-old girl with progressive limb deformities since the age of two. Upon observation the patient presented with enamel defects, absent superior incisive teeth, genu valgum, severe tibial curvature, widening of metaphyseal ends and height-for-age z-score below -3. Blood analysis revealed elevated alkaline phosphatase, hypophosphatemia, borderline low serum calcium levels, low 25-hydroxyvitamin-D, high 1,25-dihydroxycholecalciferol and parathormone; low tubular reabsorption of phosphate and a low ratio of tubular maximum reabsorption of phosphate to glomerular filtration rate. Radiographs showed metaphyseal widening, diaphyseal trabeculation, rarefaction and pseudofractures. She started support treatment and underwent orthopedic surgery with partial improvement in growth. At the age of eight, a novel heterozygous PHEX mutation (c.156_174del) was identified and she started burosumab with improvement.
In this case, there was a significant delay in the genetic diagnosis and appropriate treatment, therefore prompt referral is essential to improve the prognosis. The type of mutation may be associated with the clinical presentation, however, other factors, such as coexisting vitamin D deficiency, can explain this case severity. | | | | Keywords | | Rickets, X-Linked hypophosphatemia, burosumab. | | | | Introduction | Rickets is a common disease worldwide, characterized by a disorder of the phospho-calcium metabolism, causing deficient mineralization at the growth plates with poor growth and bone deformities. The main causes can be categorized into nutritional rickets (vitamin D, calcium and/or phosphate deficiencies) and heritable rickets (due to mutations in genes encoding proteins responsible for vitamin D activation or function, phosphate homeostasis or bone mineralization).1,2
Nutritional rickets (particularly vitamin D deficiency) remains the most frequent cause of rickets, especially in developing countries.3
Hereditary causes of rickets account for 13% of cases and include vitamin D-dependent rickets (mutations in the vitamin D receptor or vitamin D biosynthesis enzymes) or hypophosphatemic rickets (mutations in phosphatonins or phosphate transporters and co-transporters, leading to impaired renal tubular phosphate reabsorption).3
X-linked hypophosphatemia (XLH) is caused by mutations in the PHEX (phosphate regulating endopeptidase homolog X-linked) gene. It is the most common cause of genetic rickets, with an estimated incidence of 3.9 per 100,000 live births and a prevalence of 4.8 per 100,000 persons.4,5,6 The loss of PHEX function results in upregulation of FGF23 expression, leading to renal phosphate wasting, with consequent hypophosphatemia and down-regulation of 1 alpha-hydroxylase activity, resulting in decreased synthesis of active vitamin D (1,25-dihydroxycholecalciferol (1,25(OH)2D) or calcitriol). This hypophosphatemia does not respond to cholecalciferol supplementation and leads to bone mineralization defects including rickets, osteomalacia, odontomalacia and short stature.3,4,6,7
Hypophosphatemia, decreased synthesis of 1,25(OH)2D and normal serum calcium are typical laboratory findings.3,6 Serum parathyroid hormone (PTH) may be normal or slightly elevated and serum alkaline phosphatase activity is increased in children, but can be normal in adults.6
Conventional therapy for XLH consists of oral phosphate salts and calcitriol, leading to transient increase in serum phosphorus. However, normalization of serum phosphate is not achieved, leading to incomplete regression of rickets, with residual bone deformities and persistent short stature. Oral supplementation is also associated with gastrointestinal side effects, hypercalciuria, nephrocalcinosis and hyperparathyroidism.8,9
Burosumab is a recombinant human IgG1 monoclonal antibody that targets FGF-23, used for XLH treatment in patients aged ≥ 1 year. It reduces the severity of rickets with a favorable safety profile. Improving renal tubular phosphate reabsorption and increasing serum phosphorus concentration, burosumab has a positive impact on linear growth and physical function, also contributing to reduce pain.8,9,10
We report a novel PHEX mutation causing a severe clinical presentation of XLH.
| | | | Case Report | A five-year-old girl from Guinea-Bissau with progressive limb deformities was transferred to Portugal for diagnostic workup and treatment. Upper and lower limb deformities had first appeared when she started crawling at the age of 12 months and when she started walking at the age of 20 months, respectively. First tooth eruption occurred at 18 months. Canine and incisors shed at the age of four years, with no growth of permanent teeth afterward. She had no relevant family history or history of consanguinity.
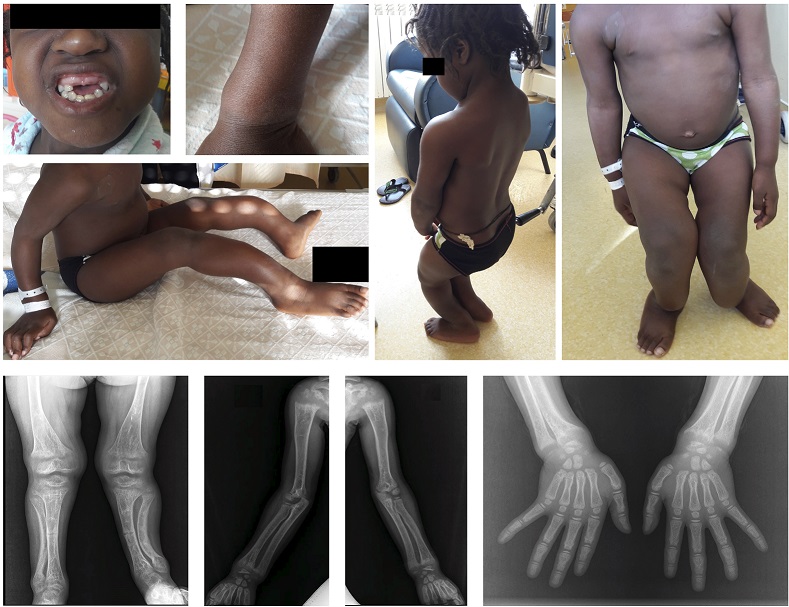
Upon observation, the patient presented with enamel defects, absent superior incisive teeth, genu valgum, severe tibial curvature in the sagittal plane, widening of metaphyseal ends (Figure 1), height-for-age z-score below -3, weight-for-age z-score between -1 and 0 and weight-for-height z-score between 2 and 3.
Figure 1. Photographs at the time of the diagnosis: absent superior incisive teeth, widening of metaphyseal ends, severe tibial curvature and genu valgum. Radiological findings at the time of the diagnosis: diaphyseal trabeculation, rarefaction and pseudofractures and metaphyseal widening.
Blood analysis (Table 1) showed elevated alkaline phosphatase (1173 U/L), hypophosphatemia (2.8 mg/dL); borderline hypocalcemia (total calcium 9.1 mg/dL; ionized calcium 1.08 mmol/L); low 25-hydroxyvitamin-D (18 ng/mL); high 1,25(OH)2D (124.18 pg/mL); and high PTH levels (212.3 pg/mL). The patient had renal phosphate wasting, as reflect by low fractional tubular reabsorption of phosphate (TRP) (57%) and low ratio of tubular maximum reabsorption of phosphate to glomerular filtration rate (TmP/GFR) (2.22 mg/dL) (Table 1). Urinalysis was normal (pH 7, density 1.020, negative for protein, glucose, leukocytes and erythrocytes) and urine calcium/urine creatinine ratio was also normal (0.16 mg/mg).
Radiographs revealed metaphyseal widening, diaphyseal trabeculation, bone rarefaction and pseudofractures (Figure 1).
Clinical, analytic and imaging findings raised the suspicion of hypophosphatemic rickets due to vitamin D-resistant rickets associated with nutritional vitamin D deficiency.
Table 1. Laboratory work-up at the diagnosis and after initiation of conventional and targeted therapy.
| |
At diagnosis |
At 13 months of conventional therapy |
Before initiation of burosumab |
12 months after burosumab initiation |
Reference values* |
| Total calcium (mg/dL) |
9.1 |
8.8 |
9.8 |
8.7 |
8.8-10-8 |
| Ca2+ (mmol/L) |
1.08 |
- |
- |
- |
1,15 – 1,32 |
| Phosphorus (mg/dL) |
2.8 |
3.6 |
2.4 |
4 |
4.1-5.9 |
| Alkaline phosphatase (U/L) |
1173 |
638 |
528 |
479 |
156-369 |
| 25-hydroxyvitamin-D (ng/mL) |
18 |
- |
- |
- |
Deficiency: < 10
Insufficiency: 10 - 30
Sufficient: 30 - 100 |
| 1,25(OH)2D (pg/mL) |
124.18 |
- |
- |
- |
19.6-54.3 |
| PTH (pg/mL) |
212.3 [16.23-63.02] |
244.2 [16.23-63.02] |
40.7 [16.23-63.02] |
115.6 [21.89–87.55] |
* |
| TRP (%) |
57 |
- |
- |
94.34 |
=85 |
| TmP/GFR (mg/dL) |
2.22 |
- |
- |
4.84 |
3.00-5.08 |
Ca 2+: ionized calcium; 1,25(OH)2D: 25-hydroxyvitamin-D; PTH: Parathormone; TRP: tubular reabsorption of phosphate;
TmP: tubular maximum reabsorption of phosphate; GFR: glomerular filtration rate
* PTH references values vary according to the patient’s age and are presented in brackets.
The patient was hospitalized and started on intravenous phosphate supplementation, showing improvement in phosphate levels six hours after treatment initiation. At day two she was started on oral phosphate supplementation and alfacalcidol. She was discharged home after fourteen days in the hospital, with stable serum phosphate levels, but no normalization.
The patient was referred to the Pediatric Nephrology, Endocrinology and Orthopedic outpatient clinics. Treatment with phosphate supplementation (45 mg/kg/day), cholecalciferol (667 IU/day) and alfacalcidol (0.5 mcg/day) was maintained. In outpatient clinic appointments, she was started on calcium supplementation (500 mg/day) due to persistent hypocalcemia (total calcium 8.7 mg/dL).
The patient underwent two orthopedic surgeries, with partial improvement of the genu valgum. In the first surgery (at the age of 5 years and 10 months) a shortening osteotomy of the left fibula and a corrective osteotomy with shortening of the tibia and internal fixation were performed. In the second surgery (at the age of 8 years and two months) a bilateral proximal tibial hemiepiphysiodesis was performed.
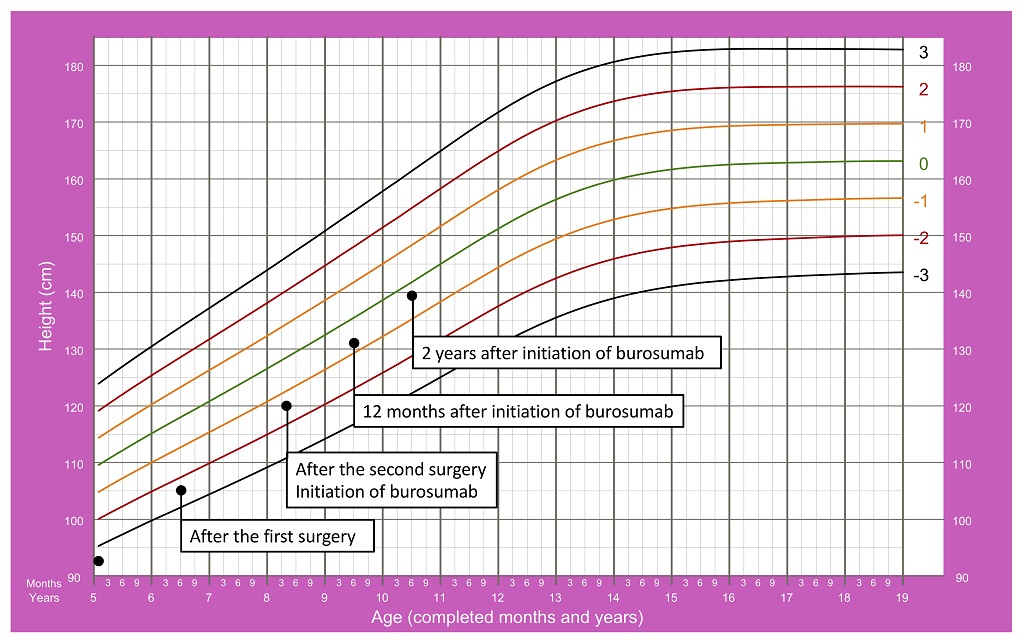
Both conventional and surgical treatments resulted in improvement in growth. At the age of eight years and three months, her height for age z-score was between -2 and -1 (Figure 2).
Figure 2. Patient’s growth chart since the admission date until 2 years after initiating burosumab.15
At the age of eight and four months, next generation sequencing identified a novel heterozygous PHEX mutation (c.156_174del). The diagnosis of XLH associated with nutritional vitamin D deficiency was established.
The patient started receiving treatment with burosumab (0.8 mg/kg every two weeks).
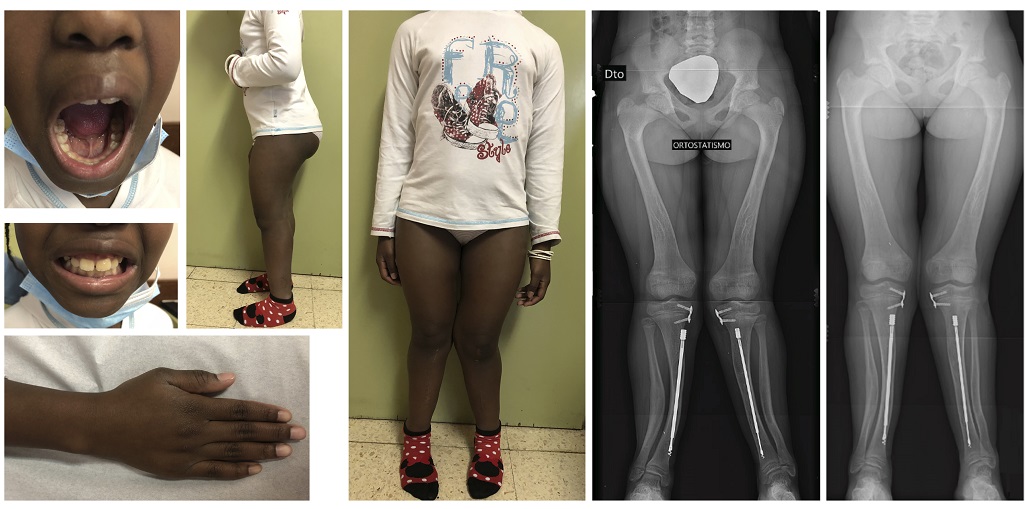
At present, she is ten years old and continues to be under treatment with burosumab, showing a major clinical response with no side effects. Remarkably, she has shown an increase in height for age (z-score between -1 and 0, as shown in Figure 2), normalization of serum phosphorus (Table 1) and improvement of enamel defects, bone deformities and bone mineralization (Figure 3).
Figure 3. Photographs seven months after the initiation of burosumab, showing clinical improvement comparing with the images shown in Figure 1. Radiographs six months (left) and one year (right) after the initiation of burosumab.
| | | | Discussion | In this case, a late referral, three years after the diagnosis of bone deformities, caused a significant delay in the genetic diagnosis and appropriate treatment initiation.
XLH is characterized by PHEX loss of function, causing an increase in FGF23, which inhibits phosphate reabsorption and the production of 1,25(OH)2D in the renal proximal tubule.11 Consequent hypophosphatemia and 1,25(OH)2D deficiency result in rickets, osteomalacia and impaired growth, as presented in our case report.
The patient we presented showed the typical clinical, radiographic and laboratory findings of XLH (such as hypophosphatemia and low TmP/GFR).10 Elevation of alkaline phosphatase, PTH and 1,25(OH)2D could be explained by hereditary vitamin D-resistant rickets or vitamin D deficiency2,3, although calcium levels were normal and PTH was only mildly elevated. Usually, in XLH, 1,25(OH)2D levels are normal or decreased.5,10 In the case we presented, 1,25(OH)2D levels were high, which could be explained by a coexisting vitamin D deficiency. The clinical picture of nutritional rickets is usually milder than seen in the case we described.2 We believe that the superimposed vitamin D deficiency may have contributed to the severe clinical manifestations observed in our patient.
Genetic testing was essential to clarify the diagnosis and to allow for the beginning of targeted therapy. A novel heterozygous truncating mutation in PHEX gene (c.156_174del) was identified and we propose that this truncating mutation might have been the key for the severity of the clinical presentation. However, the correlation between truncating mutations and a more severe phenotype in XLH is a controversial topic. Zheng et al12 studied the clinical features and molecular genetic basis of 53 pediatric patients with XLH and suggested that the truncating and non-truncating variants in PHEX gene share an identical phenotype. Other studies13,14 support a correlation between genotype and phenotype, as patients with a truncating mutation and a family history of XLH present with lower TRP, lower 1,25(OH)2D serum levels13 and more severe skeletal disease.14
Pediatric trials have shown that burosumab can improve clinical outcomes beyond conventional therapy. Accordingly, in our patient, a marked improvement in phosphorus homeostasis, enamel defects, bone deformities and growth was noticed. | | | | Conclusion | | In conclusion, prompt referral of patients with suspected XLH is essential for timely initiation of appropriate treatment to improve the prognosis. The type of mutation implicated may be associated with the clinical presentation but other factors that potentially play a role in the severity of the disease, such as vitamin D deficiency, must also be considered and corrected. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest | | The authors declare that there are no conflicts of interest. | | |
- Carpenter TO, Shaw NJ, Portale AA, Ward LM, Abrams SA, Pettifor JM. Rickets. Nat Rev Dis Primers. 2017;3:17101. [CrossRef] [PubMed]
- Chanchlani R, Nemer P, Sinha R, Nemer L, Krishnappa V, Sochett E, et al. An Overview of Rickets in Children. Kidney Int Rep. 2020;5(7):980-990. [CrossRef] [PubMed] [PMC free article]
- Acar S, Demir K, Shi Y. Genetic Causes of Rickets. J Clin Res Pediatr Endocrinol. 2017;9(Suppl 2):88-105. [CrossRef] [PubMed] [PMC free article]
- Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019;15(7):435-455. [CrossRef] [PubMed] [PMC free article]
- Trombetti A, Al-Daghri N, Brandi ML, Cannata-Andía JB, Cavalier E, Chandran M, et al. Interdisciplinary management of FGF23-related phosphate wasting syndromes: a Consensus Statement on the evaluation, diagnosis, and care of patients with X-linked hypophosphataemia. Nat Rev Endocrinol. 2022;18(6):366-384. [CrossRef] [PubMed]
- Baroncelli GI, Mora S. X-Linked Hypophosphatemic Rickets: Multisystemic Disorder in Children Requiring Multidisciplinary Management. Front Endocrinol. 2021;12:688309. [CrossRef] [PubMed] [PMC free article]
- Schindeler A, Biggin A, Munns CF. Clinical Evidence for the Benefits of Burosumab Therapy for X-Linked Hypophosphatemia (XLH) and Other Conditions in Adults and Children. Front Endocrinol . 2020;11:338. [CrossRef] [PubMed] [PMC free article]
- Carpenter TO, Whyte MP, Imel EA, Boot AM, Högler W, Linglart A, et al. Burosumab Therapy in Children with X-Linked Hypophosphatemia. N Engl J Med. 2018;378(21):1987-1998. [CrossRef] [PubMed]
- Imel EA, Glorieux FH, Whyte MP, Munns CF, Ward LM, Nilsson O, et al. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet. 2019;393(10189):2416-2427. [CrossRef] [PubMed]
- Laurent MR, De Schepper J, Trouet D, Godefroid N, Boros E, Heinrichs C, et al. Consensus Recommendations for the Diagnosis and Management of X-Linked Hypophosphatemia in Belgium. Front Endocrinol. 2021;12:641543. [CrossRef] [PubMed] [PMC free article]
- Balani S, Perwad F. Burosumab in X-linked hypophosphatemia and perspective for chronic kidney disease. Curr Opin Nephrol Hypertens. 2020;29(5):531-536. [CrossRef] [PubMed]
- Zheng B, Wang C, Chen Q, Che R, Sha Y, Zhao F, et al. Functional Characterization of PHEX Gene Variants in Children With X-Linked Hypophosphatemic Rickets Shows No Evidence of Genotype-Phenotype Correlation. J Bone Miner Res. 2020;35(9):1718-1725. [CrossRef] [PubMed]
- Morey M, Castro-Feijóo L, Barreiro J, Cabanas P, Pombo M, Gil M, et al. Genetic diagnosis of X-linked dominant Hypophosphatemic Rickets in a cohort study: tubular reabsorption of phosphate and 1,25(OH)2D serum levels are associated with PHEX mutation type. BMC Med Genet. 2011;12:116. [CrossRef] [PubMed] [PMC free article]
- Holm IA, Nelson AE, Robinson BG, Mason RS, Marsh DJ, Cowell CT, et al. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. 2001;86(8):3889-3899. [CrossRef] [PubMed]
- Centers for Disease Control and Prevention, National Center for Health Statistics. CDC growth charts: United States. http://www.cdc.gov/growthcharts/. 2000.
DOI: https://doi.org/10.7199/ped.oncall.2023.38
|
| Cite this article as: | | Borges M A, Costa M, Baptista R B, Fitas A L, Francisco T, Abranches M. A case of severe X-linked hypophosphatemia caused by a novel PHEX mutation. Pediatr Oncall J. 2023;20: 90-94. doi: 10.7199/ped.oncall.2023.38 |
|