Rakesh Kumar.
Department of Pediatrics, Katihar Medical College, Katihar, Bihar, India.
ADDRESS FOR CORRESPONDENCE
Dr. Rakesh Kumar, Assistant professor, Department of Pediatrics, Katihar Medical College, Katihar, Bihar, India.
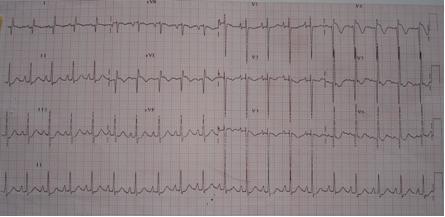
Email: drjaiswalrakesh@yahoo.co.in | | Keywords | | Coronary artery anomaly, myocardial ischemia, dilated cardiomyopathy | | | A 3 years old girl presented with breathlessness, irritability, and intermittent episodes of pallor and pain abdomen of three weeks duration. Physical examination revealed tachycardia (heart rate =150/min), tachypnea (respiratory rate = 44/min) and intercostal retractions. Peripheral pulses were weak but equal. There was cardiomegaly. Heart sounds were normal with no murmur. Liver was palpable 4cm below the right costal margin. Other systems were normal. Chest x-ray showed cardiomegaly with enlargement of left ventricle and prominent pulmonary arterial markings. Echocardiography done outside showed hypokinesia and dilatation of left ventricle (LV), ejection fraction of 30% and mild mitral regurgitation suggestive of dilated cardiomyopathy (DCM). A twelve-lead electrocardiogram (ECG) was obtained that showed anterolateral infarction with abnormal deep and wide Q waves in leads 1, aVL and V4-6 and elevated ST segment and inverted T wave in precordial leads (Figure 1). ECG was characteristic of anomalous origin of left coronary artery from pulmonary artery (ALCAPA). A repeat echocardiography was performed to trace the origin of the left coronary artery. The parasternal short axis view identified the left coronary artery arising clearly from the pulmonary artery just distal to the pulmonary valve. This established the diagnosis of ALCAPA. The child was put on anticongestive measures and after initial stabilization, referred for definitive surgical procedure.
Figure 1: ECG shows abnormal deep and wide Q waves in leads 1, aVL and V4-6 and elevated ST segment and inverted T wave in precordial leads
ALCAPA is a rare congenital malformation, with an estimated incidence of one in 300,000 live births accounting for 0.5% of congenital heart malformations. (1) First description of ALCAPA dates back to 1886 when John Brooks reported two cases of this anomaly. (1) Bland et al. in 1933 were first to correlate its clinical and autopsy findings. (2) Since then the names of Bland, White and Garland remain as eponyms. The anomaly is silent in the fetal and neonatal period when there is high pulmonary vascular resistance and pulmonary artery pressure (PAP) maintaining antegrade flow from pulmonary artery (PA) to anomalous left coronary artery (LCA). (1) With maturation of pulmonary vascular bed, decrease in PAP occurs resulting in flow reversal from LCA into PA. During this crucial transition, myocardial ischemia is an obvious sequel unless adequate intercoronary anastomosis is established between right and left coronary artery. Efficiency of this collateral circulation may be significantly jeopardized if the flow from RCA to the low resistance PA bypasses the myocardial distribution of LCA resulting in coronary steal. (3) This transition usually occurs at the age of 4-8 weeks after which 80-90% of infants born with this anomaly suffer myocardial ischemia and infarction, out of which 65-85% die in their first year of life. Survival beyond infancy, as in our case, could be due to favourable hemodynamics in the form of increased PAP, large collateral flow, narrowing of the junction between anomalous LCA and PA or right coronary artery preponderances. (3) Chronic left ventricular hypoperfusion leads to LV dysfunction and dilatation and mitral regurgitation, simulating dilated cardiomyopathy. Typically ALCAPA manifests as heart failure or angina like episodes, described as sudden onset of distressed state with ashen gray pallor, precipitated by feeding or crying and frequently misinterpreted as colics, or cardiomyopathy in infancy and rarely in early childhood, like the presentation of our case.
Diagnosis of ALCAPA requires high index of suspicion, especially in work up of cases with clinical picture of DCM in which presence of anterolateral infarction pattern on ECG together with the visualization of abnormal origin of LCA by echocardiography reasonably establishes the diagnosis. (4,5) Deep and wide Q wave in leads I and aVL and absence of wave in leads II, III and aVF are classical ECG features of ALCAPA. (4,5) Uncertainty justifies use of more sophisticated and invasive tools like coronary angiography or aortography which is considered as the definitive diagnostic procedure. Medical management includes therapy for heart failure and control of myocardial ischemia. Surgical correction is the gold standard in the therapy of ALCAPA. Simple ligation of the anomalous LCA at its origin may occasionally be required in most critically ill infants. Re-establishment of the two coronary systems by reimplanting or grafting is now considered as the procedure of choice. (6) Death from this lethal anomaly could be prevented only by means of timely diagnosis and surgical intervention; the former could be achieved by performing an ECG as a routine test in each baby around four to eight weeks of age. This will also detect some other life threatening cardiac conditions like long QT syndrome and coarctation of aorta. (7) Once detected, these conditions, lethal without timely treatment can be successfully treated in most cases. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Matherne GP, Lim DS. Congenital anomalies of the coronary vessels and the aortic root. In: Allen HD, Driscoll DJ, Shaddy RE, Feltes TF (editors). Moss and Adam's Heart disease in infants, children and adolescents, 7th edition. Philadelphia, Lippincott Williams & Wilkins 2008: 702-714.
- Bland EF, White PD, Garland J. Congenital anomalies of the coronary arteries: Report of an unusual case associated with cardiac hypertrophy. Am Heart J 1933; 8: 787-801. [CrossRef]
- Wesselhoeft H, Fawcett JS, Johnson AL. Anomalous origin of the left coronary artery from the pulmonary trunk. Its clinical spectrum, pathology and pathophysiology, based on a review of 140 cases with seven further cases. Circulation 1968; 38: 403-425. [CrossRef]
- Johnsrude CL, Perry JC, Cechin F, Smith EO, Fraley K, Friedman RA, et al. Differentiating anomalous left main coronary artery originating from the pulmonary artery in infants from myocarditis and dilated cardiomyopathy by electrocardiogram. Am J Cardiol 1995; 75: 71-74. [CrossRef]
- Chang RR, Allada V. Electrocardiographic and echocardiographic features that distinguish anomalous origin of left coronary artery from pulmonary artery from idiopathic dilated cardiomyopathy. Pediatr Cardiol 2001; 22: 3-10. [CrossRef]
- Dodge-Khatami A, Mavroudis C, Backer CL. Anomalous origin of the left coronary artery from the pulmonary artery: collective review of surgical therapy. Ann Thorac Surg 2002; 74: 946-955. [CrossRef]
- Quaglini S, Rognoni C, Spazzolini C, Priori SG, Mannarino S, Schwartz PJ. Cost-effectiveness of neonatal ECG screening for the Long QT Syndrome. Eur Heart J 2006; 27: 1824-1832. [CrossRef]
DOI: https://doi.org/10.7199/ped.oncall.2012.49
|
| Cite this article as: | | Kumar R. A CASE OF ALCAPA MASQUERADING AS DILATED CARDIOMYOPATHY. Pediatr Oncall J. 2012;9: 77-78. doi: 10.7199/ped.oncall.2012.49 |
|