Muhittin Çelik1, Mehmet Ali Kopan2, Mehmet Keskin3, Dila Tuğçe Kopan2.
1Department of Pediatrics, Division of Neonatology, Faculty of Medicine, Gaziantep University, Turkey,
2Department of Pediatrics, Faculty of Medicine, Gaziantep University, Turkey,
3Department of Pediatrics, Division of Pediatric Endocrinology and Metabolism, Faculty of Medicine, Gaziantep University, Turkey.
ADDRESS FOR CORRESPONDENCE
Mehmet Ali Kopan, Gaziantep University Faculty of Medicine Department of Pediatric Endocrinology and Metabolism, 90, Turkey.
Email: malikopan@hotmail.com | | Abstract | Propionic acidemia (PPA) is an autosomal recessive metabolic disorder caused by propionyl-CoA carboxylase deficiency, leading to accumulation of propionic acid and severe metabolic disturbances. Early recognition and management are crucial to prevent life-threatening complications.
We report a 30-day-old male neonate born to consanguineous parents who presented with feeding difficulties, vomiting, lethargy, and convulsions. Laboratory evaluation revealed pancytopenia and severe hyperammonemia. Metabolic workup, including plasma amino acids, tandem mass spectrometry, and urinary organic acids, suggested PPA, which was confirmed by a homozygous c.2003G>A mutation. The patient underwent emergency continuous venovenous haemodiafiltration (CVVHDF), resulting in rapid ammonia reduction, followed by targeted metabolic therapy including carglumic acid, l-carnitine, biotin, and metronidazole. The patient showed clinical improvement and was discharged on the 21st day.
In countries with high rates of consanguinity, organic acidemia should be suspected in neonates who develop lethargy, feeding difficulties, and convulsions within the first days of life, particularly with hyperammonemia. Hemodiafiltration can be a life-saving intervention in severe metabolic crises.
| | | | Keywords | | Propionic acidemia, Neonate, Hyperammonemia, Continuous Venovenous Haemodiafiltration (CVVHDF), Inborn errors of metabolism | | | | Introduction | Organic acidemias are inherited metabolic disorders characterized by increased urinary excretion of organic acids due to deficiencies in enzymes involved in amino acid metabolism, fatty acid β-oxidation, or carbohydrate metabolism. Propionic acidemia(PPA) is an autosomal recessive disorder affecting the catabolism of valine, isoleucine, methionine, threonine, odd-chain fatty acids, and cholesterol.1 It results from a deficiency of propionyl-CoA carboxylase, an enzyme that converts propionyl-CoA to methylmalonyl-CoA, causing the accumulation of propionic acid and clinical manifestations.2 The incidence in Turkey is 1 in 50,000–100,000.3 The enzyme comprises α and β subunits encoded by genes on chromosomes 13q32 and 3q13.3, with multiple mutations identified in affected patients.4
This case highlights the importance of considering congenital metabolic disorders in neonates with hypotonia, poor feeding, and seizures, especially in regions with high consanguinity, and the critical role of hemodialysis in managing hyperammonemia and pancytopenia. | | | | Case Report | A 30-day-old male infant, born at 38 weeks via cesarean section with a birth weight of 2950g, was admitted to the Neonatal Intensive Care Unit at Gaziantep University due to feeding difficulties, vomiting, and lethargy. The infant’s parents were healthy with second-degree consanguinity; the mother had a history of one abortion and gestational diabetes. Family history revealed involuntary, repetitive arm movements.
Patient had a high forehead, broad nasal bridge, long philtrum, epicanthal folds, hypotonia, and lethargy. The liver was palpable 5 cm below the costal margin, and primitive reflexes were absent. Laboratory tests revealed pancytopenia, and elevated serum ammonia(1096 μg/dL, reference: 68-136 μg/dL). Organic acidemia(OA) was suspected, and metabolic investigations were planned. Feeding was discontinued to halt protein intake, and high-glucose(10-12 mg/kg/day) and lipid(1 g/kg/day) infusions were started. The patient received carglumic acid(250 mg/kg loading dose, 100 mg/kg/day maintenance), carnitine(100-200 mg/kg/day), sodium benzoate(250 mg/kg/day), and metronidazole for 10 days to suppress ammonia production.
A 6.5F double-lumen hemodialysis catheter was inserted via the left internal jugular vein for emergency hyperammonemia treatment using the Seldinger technique. Haemodiafiltration was performed with a Gambro Prismaflex device and HF20 filter. Packed red blood cells primed the blood circuit, and standard solutions were used as dialysate and replacement fluids. Unfractionated heparin was given continuously at 15 IU/kg/h with aPTT monitoring every 6 hours. CVVHDF parameters were: blood flow rate 10 mL/kg/min, replacement fluid flow rate 30-50 mL/kg/h, and dialysate flow rate 2000 mL/1.73 m²/h. Hypotension was managed with dopamine at 10 mcg/kg/min. Four hours later, ammonia levels decreased to 207 μg/dL, and CVVHDF was stopped. Post-treatment, ammonia levels stayed within the reference range.
Cranial ultrasonography was normal, but EEG showed sharp slow-wave discharges at 2-Hz in the left temporal region, leading to phenobarbital initiation(5 mg/kg/day). Brain MRI revealed delayed myelination, white matter edema, effaced sulci, and cerebellar vermis atrophy with restricted diffusion in the occipital and cerebellar regions and corpus callosum.
Metabolic screening showed elevated glycine(1088 mmol/mol), increased C3-propionylcarnitine(15.3 μg/L), and elevated 2-ketoglutaric(298.8 mmol/molcreatinine) and succinic acids(162 mmol/molcreatinine). Serum methylmalonic acid was 2.05 μmol/L, and methylcitric acid was 60.50 μmol/L, indicating PPA. Bone marrow examination in pancytopenia showed normocellular to hypocellular marrow, increased promyelocytes, and vacuolization in the myeloid series.
Oral feeding was stopped to halt protein intake. High-glucose(10-12 mg/kg/day) and lipid(1 g/kg/day) infusions were initiated, along with carglumic acid (250 mg/kg loading, 100 mg/kg/day maintenance), carnitine(100-200 mg/kg/day), sodium benzoate(250 mg/kg/day), and metronidazole for 10 days to reduce ammonia production. Protein intake was reintroduced within two days, and amino acid mixtures free of precursor amino acids(Milupa OS1) were given. biotin(10 mg/day) was also administered. The patient remained stable, fed well, and had no hyperammonemia episodes. On day 21, the patient was discharged with follow-up. Genetic testing revealed a homozygous c.2003G>A mutation in the PCCA gene, confirming PPA. Genetic counseling was provided, and written consent was obtained from the parents.
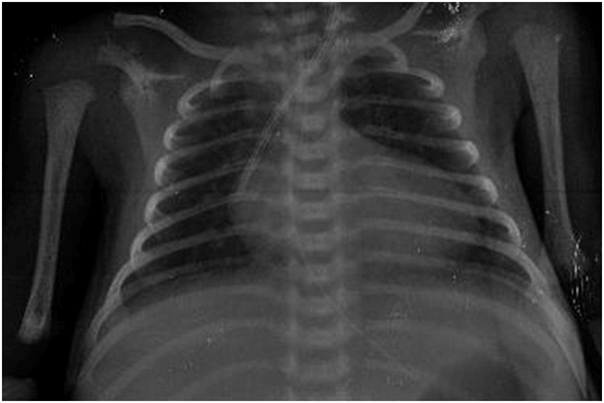
Figure 1. Chest radiograph after hemodialysis catheter placement showing the catheter position.
 | | | | Discussion | PPA is an autosomal recessive disorder affecting the catabolism of valine, isoleucine, methionine, threonine, odd-chain fatty acids, and cholesterol. Early-onset cases present with feeding difficulties, vomiting, dehydration, acidosis, hypotonia, lethargy, seizures, and coma. In our case, recurrent vomiting, hyperammonemia, feeding difficulties, lethargy, pancytopenia, and parental consanguinity suggested a metabolic disorder.
During acute attacks, metabolic acidosis, ketosis, hyperammonemia, elevated lactate, and hypoglycemia may occur. Organic acid accumulation can suppress bone marrow activity, leading to neutropenia, thrombocytopenia, or pancytopenia.2 Urinary organic acids typically show propionic acid, 3-hydroxypropionic acid, methylcitrate, tiglyl acid, and propionylglycine. Plasma methylmalonic acid may remain normal, while methylcitrate and propionylcarnitine(C3) are elevated.5 N-acetylglutamate synthase inhibition by propionyl-CoA can lead to hyperammonemia. Neuroimaging may show cerebral edema, atrophy, demyelination, and basal ganglia abnormalities, which are reversible with treatment. PPA radiological findings resemble those of methylmalonic acidemia, with mild delays in myelination.6 Diagnosis requires measurement of propionyl-CoA carboxylase activity or genetic testing.4
Acute management of PPA focuses on correcting hyperammonemia and metabolic acidosis, limiting propionate precursor metabolism through protein restriction, and providing fluids, glucose, and calories parenterally.1,2 l-carnitine(100-200 mg/kg/day) is initiated during the acute phase, and oral neomycin or metronidazole may inhibit propionic acid production. Severe hyperammonemia may require sodium benzoate, sodium phenylacetate, carglumic acid, or dialysis.7 Long-term management includes a diet restricted in valine, isoleucine, methionine, and threonine, l-carnitine supplementation, controlled protein intake (0.5-1.5 g/kg/day), and biotin(5-10 mg/day).7 Metabolic crises are triggered by protein-rich meals or infections, so prompt treatment is essential.1 Liver transplantation may be considered for severe cases.8
Hemodialysis is crucial in neonates with metabolic disorders and toxic ammonia levels, enabling rapid ammonia clearance. Studies show ammonia clearance can exceed 80% with high-flow rates and large dialyzer surfaces.9 Meta-analyses recommend hemodialysis for congenital urea cycle disorders when ammonia exceeds 500 µmol/L.9,10 Dialysis is advised for neonates and children if ammonia exceeds 500 µmol/L or if medical therapy fails within four hours. Hemodialysis is faster than peritoneal dialysis, making it more effective in acute metabolic crises.9 | | | | Conclusion | | Given the high prevalence of autosomal recessive disorders in countries with frequent consanguineous marriages, organic acidemia should be suspected in neonates who appear healthy at birth but develop lethargy, feeding difficulties, and convulsions within the first few days, especially with hyperammonemia. In patients with severe presentations requiring urgent diagnosis, like in our case, hemodiafiltration can be a life-saving intervention, enabling rapid correction of metabolic crises. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Wolf B, Hsia YE, Sweetman L, Gravel R, Harris DJ, Nyhan WL. Propionic acidemia: A clinical update. The Journal of Pediatrics. 1981 1981/12/01/;99(6):835-46. [CrossRef] [PubMed]
- De Baulny HO, Saudubray J-M. Branched-Chain Organic Acidurias. In: Fernandes J, Saudubray J-M, Van den Berghe G, editors. Inborn Metabolic Diseases: Diagnosis and Treatment. Berlin, Heidelberg: Springer Berlin Heidelberg; 2000. p. 196-212. [CrossRef]
- Tokatli A, Coskun, T. and Ozalp, I. (1993) A retrospective evaluation of 78 cases with organic acidemia. Turkish Journal of Medical Sciences, 18, 47-53.
- Kraus JP, Spector E, Venezia S, Estes P, Chiang PW, Creadon-Swindell G, et al. Mutation analysis in 54 propionic acidemia patients. Journal of Inherited Metabolic Disease. 2012;35(1):51-63. [CrossRef] [PubMed]
- Di Donato S, Rimoldi M, Garavaglia B, Uziel G. Propionylcarnitine excretion in propionic and methylmalonic acidurias: a cause of carnitine deficiency. Clinica Chimica Acta. 1984 1984/05/16/;139(1):13-21. [CrossRef] [PubMed]
- Brismar J, Ozand PT. CT and MR of the brain in disorders of the propionate and methylmalonate metabolism. American Journal of Neuroradiology. 1994;15(8):1459-73.
- Levrat V, Forest I, Fouilhoux A, Acquaviva C, Vianey-Saban C, Guffon N. Carglumic acid: an additional therapy in the treatment of organic acidurias with hyperammonemia? Orphanet Journal of Rare Diseases. 2008 2008/01/30;3(1):2. [CrossRef] [PubMed] [PMC free article]
- Vara R, Turner C, Mundy H, Heaton ND, Rela M, Mieli-Vergani G, et al. Liver transplantation for propionic acidemia in children. Liver Transplantation. 2011;17(6):661-7. [CrossRef] [PubMed]
- Cordoba J, Blei AT, Mujais S. Determinants of ammonia clearance by hemodialysis. Artif Organs. 1996;20(7):800-803. doi:10.1111/j.1525-1594.1996.tb04544.x [CrossRef] [PubMed]
- Raina R, Bedoyan JK, Lichter-Konecki U, et al. Consensus guidelines for management of hyperammonaemia in paediatric patients receiving continuous kidney replacement therapy. Nat Rev Nephrol. 2020;16(8):471-482. doi:10.1038/s41581-020-0267-8. [CrossRef] [PubMed] [PMC free article]
DOI: https://doi.org/10.7199/ped.oncall.2027.33
|
| Cite this article as: | | Çelik M, Kopan M A, Keskin M, Kopan D T. A Neonatal Case With Propionic Acidemia Undergoing Hemodiafiltration. Pediatr Oncall J. 2026 May 19. doi: 10.7199/ped.oncall.2027.33 |
|