Nooshin Baghaei, Soheila Khalilzadeh, Mohammad Reza Boloursaz, Shahin Hakimi, Amir Ali Khodayari, Ali Akbar Velayati.
National Research Institute of Tuberculosis and Lung Disease, Shahid Beheshti University of Medical Sciences and Health Services, Iran.
ADDRESS FOR CORRESPONDENCE
Amir Ali Khodayari, Masih Daneshvari Hospital, Darabad Ave, Tehran, IR Iran.
Email: dramiralikh@yahoo.com | | Abstract | Background and objectives: Cystic fibrosis (CF) is the most prevalent autosomal recessive hereditary disease in children. The mortality rate of children with CF has been reported to be higher in developing countries compared to developed countries.
This study was conducted to evaluate the clinical, paraclinical and radiological status of CF patients hospitalized in the pediatric ward of Masih Daneshvari Hospital. Complications were compared in patients who had followed their prophylactic treatment with those who did not.
Materials and Methods: Medical records of patients hospitalized at the Masih Daneshvari Hospital with a diagnosis of CF during 2000-2005 were evaluated. These patients were divided into two groups: 1) patients who had followed the prophylactic treatment regularly and 2) those who had not followed prophylactic treatment on a regular basis.
Results: A total of 32 children with CF were evaluated. Forty eight percent were males and 52% were females. The mean age of the patients was 14.46± years. The most common symptoms were chronic productive cough, developmental delay and gastrointestinal complications. The most prevalent radiological finding was bronchiectasis which was seen in 48% of the cases. The prevalence of respiratory infections and bronchiectasis was significantly higher in the second group as compared to the first (P=0.03). Five patients had passed away due to the respiratory complications of CF. The mean age of patients at the time of death was 16.5 ± 7.6 years.
Discussion and Conclusion: Occurrence of recurrent respiratory infections is one of the most distressing complications of CF. Considering the results of this study, implementing proper prophylactic treatment and training patients regarding the regular use of prescribed drugs and performing respiratory physiotherapy can be effective in decreasing disease complications.
| | | | Introduction | Cystic fibrosis (CF) is a multi-organ hereditary disorder in children and adults, which is mainly characterized by obstruction and infection of airways, gastrointestinal disorders and related consequences (1). CF is the most prevalent fatal hereditary disease of white people. The incidence has been reported to be 1 in 2000-3600 in central and Western Europe and 1 in 17000-90000 among other races (2). More than 1000 mutant genes responsible for occurrence of this disease are located on the long arm of chromosome 7. In this disease, the absorption and secretory functions characteristic of epithelial tissues are disrupted due to the involvement of CF transmembrane conductor regulator (CFTR). CFTR is a channel related to the CAMP that is found in large numbers in the epithelial cells of the airways, gastrointestinal system, sweat glands and genitourinary system (1,2,3).
Disrupted regulation of the secretions of the exocrine glands into the gastrointestinal tract can cause a wide spectrum of complications including pancreatic exocrine glands insufficiency, hepatic involvement, gastric ulcer, reflux and bowel obstruction (5).
The diagnosis of CF is made based on the positive sweat test along with symptoms of the disease. The type of pathogen responsible for respiratory disease in CF changes based on the patient's age (5). During infancy, Staphylococcus Aureus is the common pathogen and as the child grows it is replaced by Pseudomonas Aeruginosa (6). A study conducted in 2001 on 117 children with CF showed that prophylactic anti-staphylococcus antibody in affected infants- results in decreased respiratory complications in these patients (7). Several studies have demonstrated that prophylactic treatment against respiratory infections increases FEV1 and decreases the number of hospitalizations (8, 9). It appears that one of the factors responsible for progression of disease especially in Iranian patients is absence of appropriate use of prophylactic regimens.
This study aimed to evaluate the clinical, paraclinical and radiological status of patients with CF. Also disease complications were compared in patients who had followed their prophylactic treatment with those who had not.
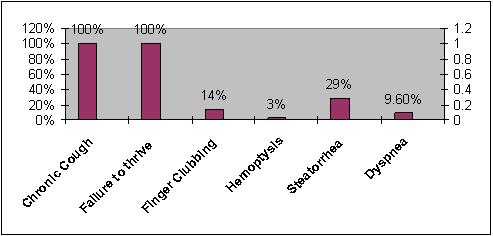
| | | | Methods & Materials | | This cross-sectional descriptive study evaluated records of CF patients during 2000-2005 at the Masih Daneshvari Hospital. All patients had positive sweat tests (CL>60 meq/L) along with clinical symptoms of the disease. Patient information including age, sex, time of diagnosis, clinical symptoms, paraclinical and radiological findings were extracted from medical files and recorded in questionnaires. The patients were divided into two groups based on their medical records: 1). Patients who had regularly followed their prophylactic regimen and 2). Those who had not followed treatment regularly. The results were analyzed by using SPSS 13 software. Tukey and T-tests were used to assess the correlation between the variables (P<0.05 was considered significant). | | | | Results | A total of 32 patients with CF were studied among which 15 patients (48%) were males and 17 (52%) were females. The mean age of the patients was 14.46 ± 6.65 yrs. The mean age of patients at the time of diagnosis was 9.5 ± 7.3 yrs. The youngest patient was diagnosed at the age of 18 months. The most prevalent clinical manifestations were chronic productive cough, delayed development and gastrointestinal symptoms (Figure 1).
Figure 1: Common clinical manifestations of Cystic Fibrosis
Chest x ray and high-resolution computed tomography (HRCT) of the lungs revealed diffuse infiltration and consolidation of both lungs in all patients. Bronchiectasis was reported in 15 patients (48%), collapse in 2 patients (5.3%) and pleural effusion in 1 patient (3.1%). We also found that 2 patients (6%) suffered from hepatic cirrhosis, 1 patient (3%) had Cor pulmonale and 2 patients (6%) had undergone appendectomy as the result of acute appendicitis. Also, 2 patients (6%) had diabetes mellitus and 1 patient (3%) had a history of pulmonary tuberculosis.
After diagnosis, prophylactic treatment comprising oral antibiotics, supplementary vitamins and respiratory physiotherapy was started for all patients. Medical records of patients disclosed that 23 patients had followed their treatment regularly while 9 patients had not. Statistical comparison between the two groups indicated that the number of respiratory infections per year was significantly lower in patients who had regular prophylactic treatment (P<0.05). Those patients that receiving prophylactic antibiotic therapy had 25-30% fewer rate of infections than other group that did not receive prophylactic therapy. Five patients (15.6%) had passed away due to respiratory complications of the disease. The mean age of these patients at the time of death was 16.5 ± 7.6 yrs.
| | | | Discussion | Cystic fibrosis was first described in 1938 by Anderson (10). CF is a disorder of exocrine gland function and multi-organ involvement results in different pathologic and clinical symptoms. The mean life expectancy of these patients is reported to be 30 years in the United States. Neonates with CF born after the year 1990 will have an average life expectancy of about 40 years (11).
In our study, the mean age of the patients at the time of diagnosis was 9.5 ± 7.3 yrs. In a similar study conducted in one of the Middle East countries the mean age of the patients at the time of diagnosis was reported to be 3.94 ± 1.81 yrs (12). Marci and colleagues in their report regarding CF in Latin America reported this age to be 7.3 ± 5.22 yrs(13).This difference may be due to the different number of understudy population or the fact that our center is a referral center for lung diseases. However, it points towards the fact that the age of diagnosing CF in our country is higher than other countries in the region.
Merquery and colleagues in the year 2000 studied the clinical symptoms of patients suffering from CF (14). Their study results are in accordance with ours. In patients with CF, progression of bronchiectasis results in focal hemorrhagic pneumonias. This is the cause of hemoptysis in CF patients (15). CT scan of the lungs of patients indicated a bronchiectatic pattern in 48% of the patients. CF is one of the main causes of bronchiectasis. Conings and colleagues in 1998 studied bronchiectasis and found that the cause of bronchiectasis in 39% of the patients was cystic fibrosis. Obstruction of intrahepatic biliary ducts in a CF patient results in biliary cirrhosis. Various studies have reported the incidence of this complication to be 15%-25% in CF patients (17, 18). In our study, 6% of the patients were suffering from hepatic cirrhosis. Accumulation of mucopurulent secretions in the airways and diffuse pulmonary damage result in cor pulmonale in CF patients (19). In our study, 3% of the patients had developed this complication. Recurrent respiratory infections in CF patients are among the most serious complications of this disease. Use of prophylactic antibiotic therapy is one of the best methods for decreasing the number of pneumonia infections in such patients (20). This study showed that proper use of prophylactic drugs can reduce the number of pneumonia infections in these patients. In a study conducted on 520 patients in the United States, a number of patients were randomly treated with tobramycin(21). Results showed that the number of hospitalizations significantly decreased in these patients. Another study on CF patients demonstrated that use of inhaled tobramycin 3 times a day improves pulmonary function and reduces the density of pseudomonas in the sputum of these patients (22).
Smyth and colleagues in 2001 revealed that prophylactic therapeutic regimens are effective for decreasing the colonization of Staphylococcus Aureus in patients less than 6 years of age (23). cephalexin is one of the most common antibiotics used for prophylactic treatment in CF patients. Stutman and coworkers in 2002 conducted a study on 209 children with CF and showed that this therapeutic regimen had no effect on the complication rate or outcome of the disease. However, it reduced the number of Staphylococcus Aureus colonies (24). Today, in many developed countries, CF patients use nebuliser drugs. Studies are indicative of the high efficacy of tobramycin nebuliser solution (TNS) method in comparison with other treatment methods (25). In our country, the majority of patients use oral antibiotics (mainly ciprofloxacin, azithromycin and cloxacillin). This can result in increased systemic complications. Considering the results of this study and those of others, proper prophylaxis, training patients in regard to the regular use of drugs especially inhaled drugs, regular respiratory physiotherapy and use of supplementary vitamins can be effective in decreasing the complications and number of hospitalizations in CF patients. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Kleigman RM, Behrman RE,.Jenson HB, Stanton BF. Nelson Text book of Pediatrics, 18th ed. Saunders. Philadelphia; 2007: 958-972.
- Kabra SK, Kabra M, Shastri S, Lodha R. Diagnosing and managing cystic fibrosis in the developing world. Paediatr Respir Rev. 2006; 7 Suppl 1:S147-50. Epub 2006 Jun 6. [CrossRef]
- Fares F, David M, Lerner A, Diukman R, Lerer I, Abeliovich D, Rivlin J. Paternal isodisomy of chromosome 7 with cystic fibrosis and overgrowth. Am J Med Genet A. 2006 Aug 15; 140(16):1785-8. [CrossRef]
- Marks JH. Airway clearance devices in cystic fibrosis. Paediatr Respir Rev. 2007 Mar; 8(1):17-23. Epub 2007 Mar 21. [CrossRef]
- Fields TM, Michel SJ, Butler CL, Kriss VM, Albers SL. Abdominal manifestations of cystic fibrosis in older children and adults. Am J Roentgenol. 2006 Nov;187(5):1199-203. [CrossRef]
- Green A, Kirk J; Guidelines Development Group. Guidelines for the performance of the sweat test for the diagnosis of cystic fibrosis. Ann Clin Biochem. 2007 Jan;44(Pt 1):25-34. [CrossRef]
- Olesen HV, Nielsen LP, Schiotz PO. Viral and atypical bacterial infections in the outpatient pediatric cystic fibrosis clinic. Pediatr Pulmonol. 2006 Dec;41(12):1197-204. [CrossRef]
- Wall M. On staphylococcal prophylaxis in CF. Pediatr Pulmonol. 2007 Feb;42(2):186. [CrossRef]
- Doron S, Gorbach SL. Probiotics: their role in the treatment and prevention of disease. Expert Rev Anti Infect Ther. 2006 Apr;4(2):261-75. [CrossRef]
- Kulczycki LL. Five decades of cystic fibrosis (1938-1988).Acta Univ Carol [Med] (Praha). 1990;36(1-4):7-12.
- Therrell BL, Lloyd-Puryear MA, Mann MY. Understanding newborn screening system issues with emphasis on cystic fibrosis screening. J Pediatr. 2005 Sep;147(3 Suppl):S6-10. [CrossRef]
- Katznelson D, Ben-Yishay M. Cystic fibrosis in Israel: clinical and genetic aspects. Isr J Med Sci. 1978 Feb;14(2):204-11. [PubMed]
- Santana MA, Matos E, do Socorro Fontoura M, Franco R, Barreto D, Lemos AC. Prevalence of pathogens in cystic fibrosis patients in Bahia, Brazil. Braz J Infect Dis. 2003 Feb;7(1):69-72. [CrossRef]
- Merqury N, Mossner J. Clinical aspects of cystic fibrosis. Med Sci Monit. 2000 Dec;11(12):RA325-8.
- Flume PA, Yankaskas JR, Ebeling M, Hulsey T, Clark LL. Massive hemoptysis in cystic fibrosis. Chest. 2005 Aug;128(2):729-38. [CrossRef]
- conings F, Grillo G, Guidi G, Rotondo A, Raia V, de Ritis G, Sarnelli P,Caterino M, Greco L. Cystic fibrosis: when should high-resolution computed tomography of the chest Be obtained- Pediatrics. 1998 May;109(5):908-13.
- Brigman C, Feranchak A. Liver Involvement in Cystic Fibrosis. Current Treat Options Gastroenterol. 2006 Dec;9(6):484-496. [CrossRef]
- Robertson MB, Choe KA, Joseph PM. Review of the abdominal manifestations of cystic fibrosis in the adult patient. Radiographics. 2006 May-Jun;26(3):679-90. [CrossRef]
- McIlwaine M. Chest physical therapy, breathing techniques and exercise in children with CF. Paediatr Respir Rev. 2007 Mar;8(1):8-16. [CrossRef]
- Harrison F. Microbial ecology of the cystic fibrosis lung. Microbiology. 2007 Apr;153(Pt 4):917-23. [CrossRef]
- Ryan G, Mukhopadhyay S, Singh M. Nebulised anti-pseudomonal antibiotics for cystic fibrosis. Cochrane Database Syst Rev. 2000;(2):CD001021. Review. Update in: Cochrane Database Syst Rev. 2003;(3).
- Ratjen F, Doring G, Nikolaizik WH. Effect of inhaled tobramycin on early Pseudomonas aeruginosa colonization in patients with cystic fibrosis. Lancet. 2001 Sep 22;358(9286):983-4. [CrossRef]
- Smyth A, Walters S. CF and antistaphylococcal prophylaxis. Thorax. 2001 Oct;56(10):819-20. [CrossRef]
- Stutman HR, Lieberman JM, Nussbaum E, Marks MI. Antibiotic prophylaxis in infants and young children with cystic fibrosis: a randomized controlled trial. J Pediatr. 2002 Mar;140(3):299-305. [CrossRef]
- Adeboyeku D, Scott S, Hodson ME. Open follow-up study of tobramycin nebuliser solution and colistin in patients with cystic fibrosis. J Cyst Fibros. 2006 Dec;5(4):261-3. [CrossRef]
|
| Cite this article as: | | Baghaei N, Khalilzadeh S, Boloursaz M R, Hakimi S, Khodayari A A, Velayati A A. A 6-year Evaluation of Cystic Fibrosis. Pediatr Oncall J. 2009;6: 23-25. |
|