Novina Novina1, Marie-José Walenkamp2.
1Child Health Department, Faculty of Medicine, Universitas Padjadjaran, Bandung, Indonesia,
2Department of Pediatrics, VU University Medical Center, Amsterdam, Netherlands.
ADDRESS FOR CORRESPONDENCE
Novina Novina, Child Health Department, Faculty of Medicine, Universitas Padjadjaran, Bandung, Indonesia
Email: novina.ade@gmail.com | | Abstract | | The most fundamental characteristics of a child are growth and development. Under normal circumstances growth follows a pattern that can be predicted. As a pediatrician we have to distinguish between normal and abnormal growth and recognize a normal variation from a pathological condition. Thorough height measurements performed every 6 months and plotted on the reference growth curve is the simplest and low-cost tool to detect an abnormal growth. Simple guideline is proposed to manage a child with short stature. | | | General introduction
The most fundamental characteristics of a child are growth and development. Under normal circumstances growth follows a pattern that can be predicted. Deviations from normal growth can be a manifestation of a wide variety of processes both endocrine disorders and non-endocrine that can involve any organ system of the body.1
General health of a child is indicated by his or her growth pattern. As a pediatrician we have to distinguish between normal and abnormal growth. We have to recognize a normal variation from a pathological condition based on medical history, growth chart, and physical examination, including body proportions and dysmorphic features. Thorough height measurements performed every 6 months and plotted on the reference growth curve is the most simple and low cost tool to recognize an abnormal growth pattern. The most important factor in evaluating growth of a child is height compared to target height.2
Growth impairment in children is indicated by some terms. Failure to thrive is defined as weight for age that falls below the 5th percentile on multiple occasions or weight deceleration that crosses two major percentile lines on a growth chart in the first years of life. Failure to thrive should not be confused with short stature or growth retardation. Failure to thrive is associated with greater impairment in weight gain than linear growth (resulting in a reduced weight-for-height ratio). Although failure to thrive may be associated with short stature or slow growth velocity, it primarily represents an inability to gain weight appropriately and only secondarily an impairment in linear growth.2 Short stature is defined as height less than -2 standard deviation (SD) or less than 2.3rd (about 3rd) percentile for a given age, sex and reference population.3,4
Great variation of congenital or acquired conditions, such as Growth Hormone Deficiency (GHD), syndromes, skeletal dysplasias, can be a cause of growth failure in industrialized countries. Meanwhile, in developing countries the major problem is malnutrition. However, other causes have to be considered hence early diagnosis and treatment of these conditions is important to prevent further health damage, to optimize adult height and increase quality of life.
Normal Growth
A complex interaction among genetic, nutritional, and hormonal factors in a cellular environment conducive for growth produces normal somatic growth. To investigate the causes of growth impairment, it is important to understand the fundamental elements of normal growth, including nutrition, oxygen, hormones, the absence of toxins, and the more general components of a healthy environment for children, including adequate sleep, exercise, and psychosocial factors. Hormonal factors, in particular, are required in the right amounts and at the right times for optimal growth. Growth hormone (GH) and insulin-like growth factor-I (IGF-I) play key roles in this process. Other hormones (e.g., thyroid hormone, insulin, sex steroids, and glucocorticoids) also affect growth, in part through their interactions with the hypothalamic-pituitary-GH-IGF axis.2,5
Intrauterine growth
Growth of a fetus starts with a single fertilized cell and ends with differentiation into more than 200 cell types, length increasing by 5000-fold, surface area by 6x106-fold, and weight by 6x1012-fold. Generally, the growth of the fetus depends on the availability of adequate oxygen and nutrition, regulated by growth factors, and supervised by an elementary genetic plan. Genetic factors are more important prior in gestation, whereas the maternal environment achieves more importance late in gestation.6
Maternal factors
Maternal factors can contribute to intrauterine growth retardation, including uterine environment such as tumors or malformations, chronic disease, chronic ingestion of alcohol or certain medications, smoking, infections as toxoplasmosis, rubella, cytomegalovirus, herpes simplex and HIV. In addition, multiple births may cause poor fetal growth.7 Maternal height is related to birth size.8
Placental factors
The placenta supplies the adequate nutrition and oxygen and regulates hormones including growth factors such as growth hormone variant, human placental lactogen and organ-specific hormones such as corticotropin-releasing hormone (CRH), hepatic and epidermal growth factors. Placental size and birth weight are positively correlated in many species. In early to mid-pregnancy, insults such as inappropriate maternal nutrition, oxygen and thermal stress can significantly disrupt placental development and subsequent fetal growth.1,6
Fetal factors
Insulin is released by the fetal pancreas as a response of nutrient reserve and is the one of the most important intrauterine growth factors. insulin-like growth factors (IGF) are produced by fetus, of which IGF-2 prevails and modifies the growth factor actions with particular binding proteins (IGFBP). Interestingly the intra uterine production of IGF-I is independent of growth hormone, in contrast to the GH dependent IGF-I secretion later in life.5
Although GH and thyroid hormone are of main importance for normal growth in childhood, their role in the control of fetal growth is relatively small, illustrated by the normal birth weight and length in most infants with congenital GH deficiency and hypothyroidism.5
Postnatal growth
Generally, the peak postnatal growth rate occurs just after birth, and decrease at mid childhood. The stunning acceleration in stature defined as the pubertal growth spurt results in a peak of growth velocity. The final deceleration in growth rate occurs until fusion of the epiphyses of the long bone then growth concludes.6
Endocrine factors
Growth hormone (GH) or somatotropin is secreted by the pituitary. The secretion is regulated by different factors. GH production is suppressed by hypothalamic GH release-inhibiting factor (somatostatin) and stimulated by GH-releasing hormone (GHRH). The effect of GH on growth is mostly mediated by IGF-I, however GH also has direct effects such as lipolysis, increased amino acid transport into tissues, and increased protein and glucose synthesis in the liver as well as some direct effects on the epiphyseal growth plate. Secretion of GH is pulsatile; The peaks of GH secretion occur in the night. Higher concentration promptly in neonatal period, decline through childhood, and incline causing increased pulse amplitude (but not frequency) during puberty stimulated by sex steroids.6
During puberty, an orchestra of sex steroid (testosterone, estrogen), GH, thyroid hormone and nutrition as instruments will accelerate growth rate known as the pubertal growth spurt. However, other factors have to be reminiscing about influence final heights. These factors include genetic, socioeconomic, psychological, physical activity, chronic disease.6,10,11
Growth monitoring
Detection of growth disorders begins with implying regular measurements of weight and length/height. Accurate and precise measurement, recording and plotting to the appropriate curves are the simplest and low cost tools. Training in measurements and plotting as well as attention to details are needed to avoid errors and imprecise plotting.12 The important point of efficient monitoring is the routine practice of measuring, recording and plotting in every office visit at least every 6 months.4
Height measurement
Every visit to a medical officer, a child should be measured his/her supine length (< 2 years of age) or standing height (> 2 years of age) as well as his/her weight. Infants must be weighed naked and children in minimal clothing. Height and growth velocity should be determined in relation to the standards for the child’s age on a graphic chart with clear indication of the child’s position (supine or standing) at measurement. Switching measurement techniques from lying to standing may falsely suggest a growth problem. Length should be measured on a firm horizontal surface with a permanently attached rule using infantometer, a stationary plate perpendicular to the rule for the head, and a movable perpendicular plate for the feet. One person should hold the head stable while another makes sure the knees are straight and the feet are firm against the movable plate. Height should be measured with the child standing back to the wall with head in frankfort horizontal position, heels at the wall, ankles together, and knees and spine straight against a vertical metal rule permanently attached to the wall or to a wide upright board. Height is measured at the top of the head by a sliding perpendicular plate (or square wooden block). A Harpenden stadiometer is a mechanical measuring device capable of such accurate measurement.6,13
Imprecise measurement can be made as a result of poor technique, variations between instruments and observers, diurnal variation, plotting mistakes also improper installation and non-calibrated tools.13
Measuring body proportions
Measurement of body proportions is important in the diagnostic assessment. In infancy the same technique and equipment used to measure length is used to measure sitting height (upper segment) but the legs are lifted vertically and the footboard brought into contact with the buttocks to measure the length of the back. After 2 years, sitting height in an ambulant child is measured by placing a seat of known height with a horizontal top under the height measuring device or using a sitting height stadiometer. Leg length (lower segment) is then calculated by subtracting sitting height (equivalent to back to head length) from standing height.1
The upper-to-lower body segment (U/L) ratio (sitting height/leg length) indicates whether the short stature is proportionate or disproportionate. The resulting U/L ratio is compared with normal values for age and sex.14
After birth the U/L ratio normally declines progressively, achieves a nadir during early puberty. When pubertal growth starts, the U/L ratio increases slightly until epiphyseal fusion. Because puberty is associated with relatively greater truncal than limb growth, an increased U/L ratio for age may be seen in precocious puberty. A decreased ratio for age indicating long legs compared to the trunk may be seen in delayed or incomplete puberty (e.g., Klinefelter or Kallmann syndrome).2,15
Arm span is obtained by measuring the fingertip-to-fingertip distance with the arms held horizontally and is an approximate representative for height (span = height 3.5 cm). Arm span can be monitored for growth velocity in children who have scoliosis, spina bifida, or leg contractures or after spinal irradiation. An abnormal relationship to height can be seen in some skeletal dysplasias or Marfan syndrome.1,2
Head circumference should be measured using a non-stretchable tape, usually as the mean of three measurements to determine the maximum occipito-frontal circumference (OFC). It may also be disproportionately large or small in some growth disorders and can be a hint for further diagnostic.1,15
Target Height
A child’s genetic potential is essential in the evaluation of his/her present growth pattern, and any deviation from the expected height for the family should raise concern. 70-80 % of height is determined by genetic factors. Many equations have been used to estimate genetic height potential. The following formulas provide an easy way of estimating the mid-parental target height (in cm) in Indonesia.4
Boys: [(mother’s height + 13) + father’s height] / 2 cm +/- 8.5 cm.
Girls: [(father’s height -13) + mother’s height] / 2 cm +/- 8.5 cm.
Parents’ heights are preferable to be measured rather than estimation or self-reports. The target height obtained by this method is then applied to the 20-year line of the gender-appropriate growth chart.12
In addition to the calculated target height, the predicted final adult height based on the skeletal age could also be considered in the evaluation of the short child. There are three methods for calculating a child’s predicted adult height, the Tanner-Whitehouse method, the Roche-Wainer-Thissen method and the Bayley-Pinneau method. However, predictions of final adult height are not very accurate and are of limited value in children with growth disorders, because the methods are based on a normal population.16,17
Growth velocity
Repeated measurement of child’s height within certain time is the most valuable base to assess growth velocity. Growth happens to be unique with small increase and not continuously linear. Growth velocity also fluctuate with the seasons, in general being fastest in spring and summer, in consequence of increased GH secretion in response to the longer periods of light.12
Growth velocity differentiates according to life stage. It is fastest in the first year of life (about 25 cm/yr overall; 38cm/yr in the first two months dropping to 12cm/ yr at one year of age), and then decreases during childhood (from 12, 10, 7, 6 and 5 cm/yr at ages 1, 2, 2–4, 4–5, and 5 years until puberty, respectively). Growth accelerates again during the pubertal growth spurt, which occurs during Tanner puberty stages II–III in girls (10 cm/yr) and Tanner IV in boys (12 cm/yr). A growth velocity of less than 5 cm/yr after age five years is disconcerting. Imprecision measurements using conventional equipment and errors in plotting on growth curve can impede accurate determination of growth velocity, therefore needs at least 3 and preferably 6 months of monitoring.2,12,18
Growth velocity (cm/year) = {[height (cm) measured at time 2 – height (cm) measured at time 1] /numbers of months between time 2 and time 1} x 12 (months per year).12
Evaluation of a child with short stature
The large number of clinical conditions associated with short stature or growth retardation can make the task of identifying a specific diagnosis particularly challenging. The evaluation of a child with short stature starts with a good medical history. Important clues can be obtained for a diagnosis as illustrated in table 1. Physical examination focuses on specific causes of short stature as summarized in table 2. If medical history and/or physical examination leads to a cause of short stature specific further diagnostic investigations follows. However, in most children that present with short stature the cause is not clear. In a child with proportionate short stature screening of secondary growth disorders (celiac disease, hypothyroidism, GH deficiency) should be performed (table 3). In disproportionate short stature a primary growth disorder is more likely and can be further investigated by a skeletal survey (table 4). In specific cases an MRI or genetic evaluation is necessary to find a cause of short stature (table 5).1,19,20
We propose an algorithm for the evaluation of a child with short stature, based on medical history, physical examination and screening laboratory investigations (Algorithm 1). The European Society of Pediatric Endocrinology (ESPE) has proposed a classification of growth disorders (table 6).
Table 1. Points to note in medical history of a child with short stature1,19
| Points |
Interpretation |
Maternal problems during pregnancy
Birth weight and length (presence or absence of ?intrauterine growth restriction)
Preterm birth, difficult birth, or breech delivery
Postnatal problems or complications
|
Small for gestational age
Syndromes
|
Family history
Parents’ and siblings’ heights, age of onset and tempo of puberty, age of attaining adult height
Medical problems (parents, siblings, grandparents)
History of consanguinity or familiar congenital anomalies
|
Constitutional short stature with pubertal delay
Growth hormone deficiencies, metabolic disorders
Syndromes, chromosomal disorders
|
Developmental milestones
School performance
|
Syndromes, chromosomal disorders, metabolic disorders
|
Medication
Topical and inhaled steroid preparations
Methylphenidate or other stimulants Anticonvulsants
Antidepressants
|
Iatrogenic causes
|
|
Fatigue, general malaise
|
Anemia, celiac disease, kidney disease
|
|
Urinary tract infections, urinary problem
|
Kidney disease
|
|
Sluggishness, constipation
|
Hypothyroid
|
Activity
Physical activity, exercise tolerance, stamina
|
Over exercise
|
|
Psychopathology
|
Emotional deprivation, depression, anorexia
|
Age of Pubertal Development
Body odor, Acne
Breast development
Genital development
Axillary and pubic hair development
Age of menarche
|
Early or delay puberty, thelarche, menarche
History of cryptorchidism
|
Medical events
Head injury, surgery, illnesses
|
Brain insult
|
Persistent symptoms
Headaches, vision or hearing problems
|
Brain tumor
|
Table 2. Points to note in physical examination of a child with short stature1,19
| Points to note |
Interpretation |
| Abnormal body proportion, abnormal upper/lower segment ratio |
Skeletal dysplasia, rickets, pseudohypoparathyroidism, osteogenesis imperfecta, mucopolysaccharidosis, mucolipidosis |
| Head circumference |
Syndromes, congenital infection, IGF-1 deficiency/resistance |
| Frontal bossing, Doll’s face |
Growth hormone deficiency |
| Fundoscopy, visual acuity |
Intracerebral process disrupting hormonal production |
Dysmorphic features: palate shape, ear placement, webbing, low hairline, size and shape of hands and feet
Chest: widely spaced nipples, pectus excavatum, shield-shaped chest
|
Syndromes, chromosomal disorders |
| Adiposity |
Cushing, Hypothyroidism, growth hormone deficiency |
| Abdominal distention |
Celiac disease |
| Enlarged Liver and/or Spleen |
Liver disease and/or Metabolic disease |
| Increased blood pressure |
Cushing, Kidney disease |
| Hirsutism |
Cushing |
| Goiter, slow pulse, slow tendon reflex |
Hypothyroidism |
| Micropenis, cryptorchidism |
Hypogonadism, hypopituitarism |
Table 3. Laboratory screening of a child with proportionate short stature1,19
| Suggested investigations |
Interpretation |
| Hemoglobin, hematocrit, leucocytes, red cell indices, leucocyte counting, ESR |
Anemia, infections |
| Urea, creatinine, sodium, potassium, calcium, phosphate, alkaline phosphatase, iron, ferritin, albumin, liver function |
Renal disorders, Metabolic bone disease, malabsorption, liver disorders |
| Acid–base status |
Renal Tubular acidosis, |
| Anti-endomysium antibodies (EMA), Anti-transglutaminase antibodies (ATA) |
Celiac disease |
| TSH, Free T4 |
Hypothyroidism |
| Insulin-like Growth Factor-1 |
Growth hormone deficiency |
| Urine analysis, (simple biochemistry and microscopy) |
Diabetes, Kidney disease. |
| Chromosomal analysis* |
Turner syndrome (*If no cause for the short stature in girls of all ages is found, a karyotype should be done) |
| Bone age |
Skeletal age, abnormality of phalanges, Madelung deformity, syndromes |
Table 4. Imaging in disproportionate short children1,19
| Suggested investigation |
Interpretation |
| Bone survey: |
Skeletal dysplasia |
| - Skull (PA and lateral) |
Craniosynostosis, shape |
| - Spine and ribs (AP and lateral) |
Scoliosis, kyphosis, lordosis
Abnormality of Spinal |
| - Thorax (AP) |
Abnormality of the ribs, scapula and clavicles |
| - Pelvis (AP) |
Abnormality of iliac wings |
| - Long bones= 1 arm and 1 leg (AP) |
Differentiation between rhizomelic and acromelic shortening |
| -Left hand (PA) |
Abnormality of the metacarpals and digits |
Table 5. Specific investigations in a child with short stature19,20
| Suggested investigation |
Interpretation |
| MRI |
Visualization of hypothalamus, pituitary |
| Growth hormone (GH) provocation test |
Growth hormone deficiency |
| Indicated genetic analyses |
Genetic disorders
Genetic defects in one of the components
(pituitary GH secretion, GH receptor (GHR), post-receptor signaling and IGF-I)
|
Table 6. The ESPE classification system of growth disorders
| A. Primary growth disorders |
B. Secondary growth disorders |
C. Idiopathic short stature |
A.1 Clinically defined syndromes
A.2 Small for gestational age with failure of catch-up growth
A.3 Skeletal dysplasias
A.4 Dysplasias with defective mineralization
|
B.1 Insufficient nutrient intake (malnutrition)
B.2 Disorders in organ systems
B.3 Growth hormone deficiency (secondary IGF-1deficiency)
B.4 Other disorders of the growth hormone-IGF axis (primary IGF-
deficiency and resistance)
B.5 Other endocrine disorders
B.6 Metabolic disorders
B.7 Psychosocial
B.8 Iatrogenic
|
C.1 Familial (idiopathic) short stature
C.2 Non-familial (idiopathic) short stature
|
Adapted from: ESPE Classification of Paediatric Endocrine Diagnoses 2007.27
Algorithm 1. Algorithm for the evaluation of a child with short stature
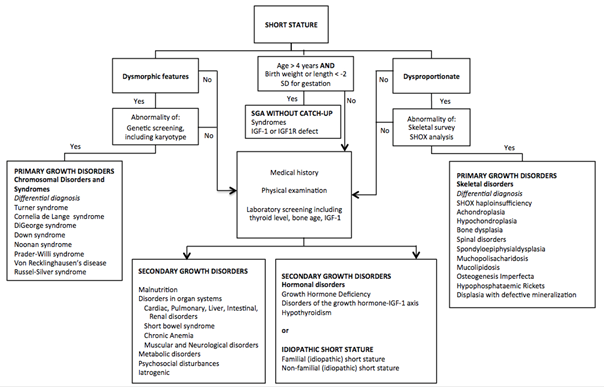
Note: SD: Standard Deviation, IGF-1: insulin-like Growth Factor-1, IGF1R: insulin-like Growth Factor-1 Receptor, SHOX: Short stature Homeobox.
Indications for GH therapy
Growth hormone deficiency is still the main reason for growth hormone replacement treatment, but nowadays GH is also being used in non-GH deficient syndromes associated with short stature. Aim of GH therapy is to normalize height in short or long term and to achieve better final height. GH is also used to improve body composition, bone density, cardiovascular risk, respiratory function, behavior, socialization and self-esteem in patients with Prader Willi syndrome.21,22,23
Non-endocrine problems of growth
Nutritional growth retardation (NGR) is most prevalent in populations at risk of poverty. However in affluent communities patients with NGR are often referred to the specialist because of short stature and delayed sexual development. Patients with NGR do not appear wasted, and the usual biochemical parameters of nutritional status, including serum levels of retinol-binding protein, pre-albumin, albumin, transferrin, and triiodothyronine (T3) levels, do not differentiate NGR patients from those with familial or constitutional short stature. The explanation is that NGR patients have adjusted to their suboptimal nutritional intake and they maintain homeostasis by decelerating growth, thereby reaching equilibrium with continuity of biochemical nutritional markers and genetic growth potential. Deceleration of growth creates the nutrient demands into balance with the nutritional intake, without adversely affecting biochemical or functional homeostatic measures.12,24
The high prevalence of infections in developing countries attenuates linear growth of children living in impoverished area. Such infections can decrease linear growth by affecting nutritional status. Whilst infection of cells directly involved in bone remodeling (osteoclasts or osteoblasts) by specific viruses may also directly affect linear growth. Acute or chronic inflammation may modulate long bone growth via induction of IL-6 production. Induction of the acute phase response and production of proinflammatory cytokines such as TNF-α, IL-1β and IL-6 may directly affect the process of bone remodelling that is required for long bone growth. TNF- α could lead to increased production of macrophages and diminished production of osteoclasts.25
Emotional deprivation can cause short stature and should be considered in all cases with unexplained growth retardation. Symptoms such as hyperphagia, abnormal eating habits, disturbed behaviour, global development delay, enuresis and encopresis especially the possibility of sexual abuse should be examined.26
| | | | Conclusion | | The management of a child with short stature is a challenge for every pediatrician. In this article we have given an overview over the process of normal intrauterine and postnatal growth. We highlighted the importance of adequate measurement of height and body proportions in order to distinguish primary and secondary growth disorders. Medical history and physical examination is essential in the evaluation of a child of short stature. We have proposed a guideline that can be used in the management of a child with short stature. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Wales JK. Evaluation of growth disorders. Brook's clinical pediatric endocrinology 6th edn, 2009: 124-54. [CrossRef]
- Rose SR, Vogiatzi MG, Copeland KC. A general pediatric approach to evaluating a short child. Pediatrics in Review 2005; 26: 410-9. [CrossRef] [PubMed]
- Wit JM, Ranke MB, Kelnar CJH. ESPE classification of pediatric endocrine diagnoses. Horm Res 2007; 68 (Suppl 2): 1–120.
- Pujiadi AH, Hegar B, Handyastuti S, Idris NS, Gandaputra EP, Harmoniati ED, editor. Pedoman Pelayanan Medis Ikatan Dokter Anak Indonesia. Available from http: idai.or.id/downloads/PPM/Buku-PPM.pdf
- Forbes K, Westwood M. The IGF axis and placental function. A mini review. Horm Res 2008; 69: 129-37. [PubMed]
- Styne D. Growth. In Gardner DG, Shoback D, editors. Greenspan`s basic and clinical endocrinology 8th ed. Philadelphia: Mc Graw Hill. 2007: 171-208.
- Roland MCP, Friis CM, Godang K, Bollerslev J, Haugen G, Henriksen T. Maternal Factors Associated with Fetal Growth and Birthweight Are Independent Determinants of Placental Weight and Exhibit Differential Effects by Fetal Sex. Open access 2014:9(issue 2). Available from http: www.plosone.org
- Yaw Addo O, Stein AD, Fall CH, Gigante DP, Guntupalli AM, Horta BL et al. Maternal height and child growth patterns. The journal of pediatrics 2013; 163 : 549-54. [CrossRef] [PubMed] [PMC free article]
- Forbes K, Westwood M. Maternal regulation of placental development. J Endocrinol. 2010;207:1-16. [CrossRef] [PubMed]
- Rogol AD, Clark, PA, Roemmich JN. Growth and pubertal development in children and adolescents: effects of diet and physical activity. Am J Clin Nutr 2000; 72(suppl): 521S-8S. [CrossRef] [PubMed]
- Melmed S, Kleinberg D, Ho K. Pituitary physiology and diagnostic evaluation. In: Melmed S, Polonsky KS, Larsen PR (eds). Williams's textbook of endocrinology, 12th edn. Saunders 2011:175-228. [CrossRef]
- Grimberg A, Lifshitz F. Worrisome growth. In: Lifshitz F (eds). Pediatric endocrinology Vol 2. 5th ed. Informa healthcare. 2007: 1-50.
- Hall DMB. Growth monitoring. Arch Dis Child 2000; 82: 10–15. [CrossRef] [PubMed] [PMC free article]
- John Hopkins Hospital. Children's medical and surgical center. The Harriet Lane handbook. 7th ed. Chicago: Year Book Medical Publishers, c1975.
- Nicol LE, Allen DB, Czernichow P, Zeitler P. Normal growth and growth disorders. In Kappy MS, Allen DB, Geffner ME, Eds. Pediatric practice Endocrinology. The McGraw-Hill co, Inc. 2010; 23-76.
- Bayley N, Pinneau SR. Tables for predicting adult height from skeletal age: revised for use with the Greulich-Pyle hand standard. J Pediatr 1952; 40: 423–441. [CrossRef]
- Greulich WW, Pyle SI. Radiographic Atlas of Skeletal Development of the Hand and Wrist. Stanford, CA: Stanford University Press, 1950. [PubMed]
- Growth velocity for the average-maturing children. Developed by the National Center for Chronic Disease Prevention and Health Promotion (2000). Available from: http: www.cdc.gov/ growth charts.
- Oostdijk W, Grote FK, Muinck Keizer-Schrama SMPF, Wit JM. Diagnostic approach in children with short stature. Horm Res 2009; 72: 206-17. [CrossRef] [PubMed]
- Walenkamp MJE, Wit JM. Genetic disorders in the growth hormone-insulin-like growth factor-I axis. Horm Res 2006; 66: 221-30. [PubMed]
- Kirk J. Indications for growth hormone therapy in children. Arch Dis Child 2012; 97: 63–68. [CrossRef] [PubMed]
- Growth Hormone Research Society. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. J Clin Endocrinol Metab 2000; 85: 3990-3. [CrossRef] [PubMed]
- Richmond E, Rogol AD. Current indications for growth hormone therapy for children and adolescents. Hindmarsh PC editor. Endocr Dev. Basel, Karger 2010; 18: 92-108.
- Lifshitz F. Nutrition and growth. J Clin Res Ped Endo 2009; 1(4): 157–163.
- Stephensen CB. Burden of infection on growth failure. 1999 Available from jn.nutrition.org
- Gohlke BC, Khadilkar VV, Skuse D, Stanhope R. Recognition of children with psychosocial short stature: a spectrum of presentation. J Pediatr Endocrinol Metab. 1998 Jul-Aug;11(4):509-17. [CrossRef] [PubMed]
- Short Stature. Wit JM, Ranke MB, Kelnar CJH, eds. ESPE Classification of Paediatric Endocrine Diagnoses. Hormone Research. Karger 2007;1-9.
DOI: https://doi.org/10.7199/ped.oncall.2019.19
|
| Cite this article as: | | Novina N, Walenkamp M. MANAGEMENT OF CHILDREN WITH SHORT STATURE. Pediatr Oncall J. 2019;16: 35-42. doi: 10.7199/ped.oncall.2019.19 |
|