Rita Sousa, Andreia Romana, Nádia Santos, Joana Jorge, Rita Marques, Margarida Pinto, Paulo Calhau.
Department of Pediatrics, Hospital Garcia de Orta, Almada, Portugal.
ADDRESS FOR CORRESPONDENCE
Rita Sousa, Avenida Torrado da Silva, 2805-267 Almada, Portugal.
Email: rita.mcosousa@gmail.com | | Abstract | Pseudohypoparathyroidism is a rare inherited cause of hypocalcemia, defined as a group of disorders with end-organ resistance to parathyroid hormone (PTH) and characterized by hypocalcemia, hyperphosphatemia and elevated PTH serum levels.
We report an uncommon case of a 17-year-old female with repeated episodes of impairment of consciousness associated with carpopedal spasm. Physical examination revealed a short stature, a rounded face and flattened nasal pyramid; laboratory evaluation showed low serum calcium level, elevated serum phosphorus levels and an elevated serum parathyroid hormone level; the ECG revealed a prolonged QTc interval. CT Brain revealed calcifications in the basal ganglia and phacosclerosis suggestive of chronic hypocalcemia. These results together with the phenotypic findings onphysical examination led to the diagnosis of Albright's Hereditary Osteodystrophy. The adolescent showed an improvement with medical management.
| | | | Keywords | | hypocalcemia, parathyroid hormone, pseudohypoparathyroidism. | | | | Introduction | Hypocalcemia may be an asymptomatic finding or there may be a wide range of correlated signs and symptoms.1 In patients who develop acute hypocalcemia, symptoms such as tetany and seizures may occur. Patients with chronic hypocalcemia may develop ectodermal and dental changes, cataracts, basal ganglia calcification and extrapyramidal disorders.1,2 There are multiple causes of hypocalcemia in pediatric age, with hypoparathyroidism and vitamin D deficiency being the most common.3 Pseudohypoparathyroidism (PHP) is a rare cause of hypocalcemia. First described in 1942 by Albright et al, the PHP refers to a group of disorders defined by end-organ resistance to PTH, specifically at the proximal renal tubule and characterized by hypocalcemia, hyperphosphatemia, elevated PTH and low vitamin D serum levels.7,8 PHP is characterized by an inadequate response to the parathyroid hormone (PTH) produced by a loss of G-protein-mediated signaling.4 This rare inherited disease has an estimated prevalence of 0.3 to 1.1/100.000 and is caused by inactivating mutations within the GNAS gene or epigenetic alterations at the GNAS locus, that are present in both sporadic or autosomal dominant heterozygous and identified in 80-90% of patients with PHP.5 The subtypes included are PHP type 1a (PHP-1a), PHP type 1b (PHP-1b), PHP type 1c (PHP-1c), PHP type 2 (PHP-2) and pseudopseudohypoparathyroidism (PPHP). In some cases, resistance to other hormones, such as TSH (PHP-1a, PHP-1c), follicle stimulating hormone and luteinizing hormone (PHP-1a, PHP-1c) or growth hormone-releasing (PHP-1a) can be observed.8,9 Symptoms of hypocalcemia may not be present at birth and resistance to PTH arises over time despite the presence of the mutation from birth.6 Biochemically this disorder is characterized by hypocalcemia, hyperphosphatemia and elevated PTH serum level. There are several subtypes of PHP, with different genetic and clinical characteristics. Some of these variants are associated with skeletal abnormalities, cognitive impairment and resistance to other hormones.5
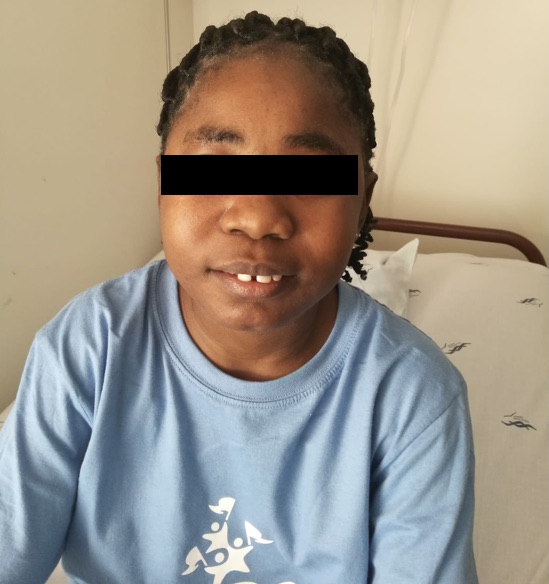
The authors present a case of an adolescent girl with severe symptomatic hypocalcemia manifested by signs of tetany and electrocardiographic changes, later leading to the diagnosis of PHP. | | | | Case Report | A 17-year-old female patient presented to the pediatric emergency department complaining of five episodes of impairment of consciousness on the admission day, lasting for a few seconds, without urinary or fecal incontinence, abnormal movements, sialorrhea, cyanosis or excessive sleepiness. These episodes were preceded by headache, tunnel vision and dizziness. She denied chest pain, palpitations, respiratory or gastrointestinal symptoms. She reported similar episodes for 2 years, monthly and worsening in the previous 3 days. Regarding her past medical history, she denied previous admissions or surgeries, as well as usual medication. No investigations of the previously described episodes were carried out in her home country. A cognitive impairment was noticed since early age and she was currently at level 2 of education according to the International Standard Classification of Education (ISCED) 201110, having failed a total of 4 years of schooling. The parents were healthy and non-consanguineous with no relevant family medical history. Physical examination revealed a short stature, with a height of 144 cm (P<3, WHO Reference 2007) and a weight of 42 Kg (P<5, WHO Reference 2007), a rounded face with lower maxillary hypoplasia and flattened nasal pyramid (Figure 1). Vital signs were normal. Otherwise, the physical examination was unremarkable. During medical observation, three episodes lasting 2 to 3 seconds of loss of contact were noted, one of them with carpopedal spasm. The laboratory evaluation carried out showed hemoglobin 13.2 g/dL [11.5–18.0], platelets 284.000 cells/μL [130-400], leukocytes 4.5x109 cells/μL [4,0-11,0] with 48,7% neutrophils [40-74], glucose 93 mg/dL [60-110], urea 22 mg/dL [16-48], creatinine 0.7 mg/dL [0.5-0.9], potassium 3,6 mmol/L [3.5-5.0], sodium 140 mmol/L [135-148], AST 36 UI/L [<32], ALT 24 UI/L [<24], C reactive protein <0,06 mg/dL [<0.2]. Venous blood gas showed pH 7.37, pCO2 52.0 mmHg, pO2 37 mmHg, HCO3 30,1 mmol/L, calcium 0,53 mmol/L [1.00-1.32], Cl- 99 mmol/L [98-106], lactate 20,00 mg/dL [<11]. The electrocardiogram revealed a normal rhythm, prolonged QTc interval (460 ms [<450 ms]) at a heart rate of 91 beats/min with a lengthening of the ST segment.
Figure 1. Head CT scan (axial view) showing multiple intracranial calcifications.
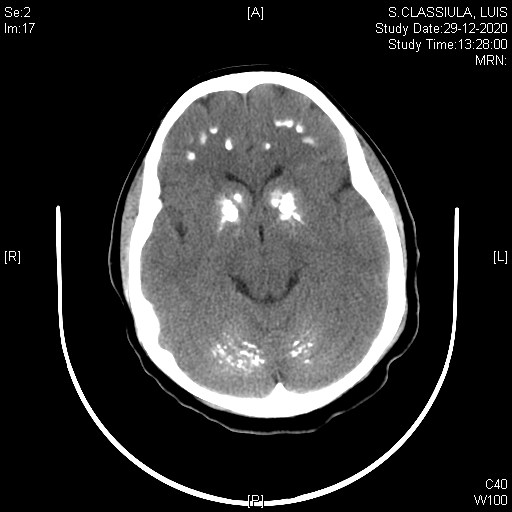
The diagnosis of a symptomatic hypocalcemia was made and we promptly initiated correction with intravenous calcium administration (3 days perfusion of calcium gluconate 2 mg/Kg/h). Laboratory investigation of hypocalcemia revealed phosphorus 8.8mg/dL [2.5-4.5], total serum calcium 5.4 mg/dL [8.1-10.2], albumin 4.5 g/dL [3.5-5.0], ALP 357 U/L [35-104], FSH 5.3 UI/L [3.5-12.5], LH 11.7 UI/L [2.40-12.6], GH 2.5 ng/mL [<10], IGF-1 199 ng/mL [190-429], cortisol 11.1 ug/dL [6.24-18], estradiol 196 pg/mL [86-498], acth 48.6 pg/mL [9-50], TSH 1.71 mU/L [0.27-4.20], free T4 1.45ng/dL [0.93-2.70], PTH 675 pg/mL [15-65], vitamin D 24.9 ng/mL [>30], urinary calcium <0.8 mg/dL [6.60-21.30]. These results increase the suspicion of PHP. In this context, a Head CT scan was carried out which revealed multiple intracranial, supra and infratentorial calcifications, polymorphous, symmetrical and of interest to the basal gray nuclei, thalamus, bilateral frontal and parietal subcortical white matter and dentate nuclei of the cerebellum (Figure 2). Ophthalmological observation showed anterior capsular phacosclerosis, assessment of hand bone age corresponded to chronological age and renal ultrasound showed no signs of nephrocalcinosis. Array comparative genomic hybridization showed a normal female pattern and the GNAS (guanine nucleotide binding protein, alpha stimulating) gene sequencing revealed no anomalies. Supplementation with calcium (1500 mg + 3000 mg per day), cholecalciferol (400 UI + 800 UI per day) and calcitriol (0,25 mcg twice a day) was implemented and the patient was referred to endocrinology and neurology consultation for further follow-up. From that time, there was a progressive normalization in calcium levels until a low-normal level, with no recurrence of the described episodes.
Figure 2. Phenotypic findings of the physical examination included a short stature, round face lower maxillary hypoplasia and flattened nasal pyramid.
 | | | | Discussion | Hypocalcemia may be associated with a spectrum of clinical manifestations ranging from asymptomatic when mild to seizures, tetany or refractory heart failure if severe.1,6 The main symptom of acute hypocalcemia is tetany, which is characterized by neuromuscular irritability and can be translated from paresthesia’s or muscle cramps to more severe symptoms such as seizures, laryngospasm and carpopedal spasm. The latter can be observed in the case of the patient described above.1 The etiology of hypocalcemia can be related to a failure in one of components of the calcium homeostatic system, such as deficiency of or resistance to PTH or vitamin D or a defect of the calcium-sensing receptor.7 PHP is characterized by hypocalcemia, hyperphosphatemia, elevated PTH and low vitamin D serum levels.7,8 All these biochemical changes can be found in the clinical case described previously. PHP presents a wide variability of clinical manifestations.9 The set of typical clinical manifestations presented by this patient is called Albright's Hereditary Osteodystrophy, including short stature, round face and cognitive impairment.9,11 Although the GNAS gene sequencing did not detect anomalies, literature describes that 10 to 20% of cases have no genetic cause identified.8
The clinical case described represents a case of Albright's Hereditary Osteodystrophy (PHP-1a) with characteristic biochemical (hypocalcemia, hyperphosphatemia with high levels of serum PTH) and phenotypic findings (short stature, round face and cognitive impairment).9 Initially our patient was managed symptomatically with intravenous calcium and posterior with oral calcium and calcitriol supplements. PHP approach should include multidisciplinary interventions and it has a good prognosis when an adequate calcium and vitamin D reposition is assured.9
The important clinical feature in our case is severe symptomatic hypocalcemia that presents with signs of tetany, electrocardiographic abnormalities and basal ganglia calcifications, later leading to the diagnosis of PHP particularly in its classic form, the Albright's Hereditary Osteodystrophy.
What this report shows
• There are multiple causes of hypocalcemia in pediatric age, with hypoparathyroidism and vitamin D deficiency being the most common.
• Pseudohypoparathyroidism is a rare cause of hypocalcemia, as a group of disorders defined by end-organ resistance to PTH and characterized by hypocalcemia, hyperphosphatemia, elevated PTH and low vitamin D serum levels.
• The typical clinical manifestations presented in patients with pseudohypoparathyroidism is called Albright's Hereditary Osteodystrophy, including short stature, round face and cognitive impairment.
• Pseudohypoparathyroidism’s approach should include multidisciplinary interventions and it has a good prognosis when an adequate calcium and vitamin D reposition is assured. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Goltzman D. Clinical manifestations of hypocalcemia. In: UpToDate, Pst TW (Ed), UpToDate, Waltham, MA. (Accessed on August, 2021).
- Dawrant J, Pacaud D. Pediatric hypocalcemia: making the diagnosis. CMAJ. 2007; 177(12): 1404-1497. [CrossRef] [PubMed] [PMC free article]
- Najim M, Ali R, Omer A. Pseudohypoparathyroidism presenting with seizures: a case report and literature review. Intractable Rare Dis Res. 2020; 9(3): 166-170. [CrossRef] [PubMed] [PMC free article]
- Mantovani G, Spada A, Elli FM. Pseudohypoparathyroidism and Gsα-cAMP-linked disorders: current view and open issues. Nature Reviews - Endocrinology. 2016; 12: 347-356 [CrossRef] [PubMed]
- Tafaj O, Jüppner H. Pseudohypoparathyroidism: one gene, several syndromes. J Endocrinol Invest. 2017;40(4):347. [CrossRef] [PubMed]
- Cooper MS, Gittoes NJL. Diagnosis and management of hypocalcaemia. BMJ. 2008; 336:1298-302. [CrossRef] [PubMed] [PMC free article]
- Carpenter T. Etiology of hypocalcemia in infants and children. In: UpToDate, Pst TW (Ed), UpToDate, Waltham, MA. (Accessed on August, 2021).
- Sinnott BP. Pseudohypoparathyroidism-Literature Update. Ann Endocrinol Metab. 2018; 2(1):26-33. [CrossRef]
- Mantovani G et al. Recommendations for Diagnosis and Treatment of Pseudohypoparathyroidism and Related Disorders: An Updated Practical Tool for Physicians and Patients. Hormone Research in Paediatrics. 2020; 93:182-196. [CrossRef] [PubMed] [PMC free article]
- United Nations Educational, Scientific and Cultural Organizations (UNESCO). International Standard Classification Education (ISCED). UNESCO Institute for Statistics. 2011.
- Martos-Moreno GA, Lecumberru B, Nanclares GP. Implicaciones en pediatría del primer consenso internacional para el diagnóstico y asistencia a pacientes com pseudohipoparatiroidismo y enfermedades relacionadas. Anales de Pediatria. 2019; 90(2):125.e1-125.e12. [CrossRef] [PubMed]
DOI: https://doi.org/10.7199/ped.oncall.2024.2
|
| Cite this article as: | | Sousa R, Romana A, Santos N, Jorge J, Marques R, Pinto M, Calhau P. A Rare Cause of Severe Hypocalcemia in Paediatrics. Pediatr Oncall J. 2024;21: 30-32. doi: 10.7199/ped.oncall.2024.2 |
|