Prashanth RR, Medha Goyal, Jyothi Sreekumari, Anitha Haribalakrishna.
Department of Neonatology, Seth G.S. Medical College and King Edward Memorial Hospital, Mumbai, India.
ADDRESS FOR CORRESPONDENCE
Prashanth RR, Department of Neonatology, Seth G.S. Medical College and King Edward Memorial Hospital, Mumbai, India.
Email: prash2635@gmail.com | | Abstract | | In this case report, we describe an uncommon presentation of choledochal cyst in a preterm infant, second of twins, born at 33+1 weeks by date, weighing 1434 g, born to an HCV positive mother. The anomaly was not identified in any antenatal ultrasound studies performed in any trimester. The clinical situation had presented us with a unique diagnostic dilemma with the first of the twins thriving well, in contrast to the second twin who developed unphysiological weight loss and neonatal cholestasis from day of life 23. Though the presentation was typically characteristic of an obstructive cause, this was overshadowed by premature status. There is a dearth of published literature regarding neonatal choledochal cyst, its presentation in preterm infants, and in twin babies. | | | | Keywords | | Neonate, Choledochal Cyst, Preterm, Twin. | | | | Introduction | | The most common variant of choledochal cyst (CC) is Type 1(80-90% of all choledochal cyst.1 They are fusiform or spherical dilatation of the extrahepatic biliary tree. All neonatal cases of choledochal cysts need early surgical intervention to prevent liver fibrosis-associated complications and to facilitate complete functional recovery. This is the first case report in literature, where we describe an uncommon presentation of Type 1c choledochal cyst in a preterm infant, second of twins, born at 33+1 weeks, weighing 1434 g, with cooccurrence of HCV infection in the mother. | | | | Case Report | A 40 years old G2P2A1L0 delivered a male neonate at 33 weeks of gestation with a birth weight of 1434 grams. Neonate had APGAR scores of 9 at both 1 and 5 minutes, and no resuscitation was required. On revisiting history neonate was born of non consanguineous marriage via vaginal route. Mother was diagnosed to be HCV positive during routine antenatal screening at 6 months of gestation. Her HCV RNA titres were 106 and HCV antibody was reactive. Her SGOT/SGPT at the time of diagnosis was 367/140 and Total Bilirubin/Direct bilirubin-0.2/0.1. Other liver functions were normal. HIV and VDRL tests were negative. She was started on oral ribavirin but defaulted treatment. The pregnancy was conceived via in vitro fertilization with embryo transfer. Antenatal sonography was suggestive of dichorionic diamniotic (DCDA) twins with normal doppler studies. She went into spontaneous preterm labour, after adequate antenatal steroid coverage she delivered the twins via vaginal route. The index neonate second of the twin, the baby boy weighing 1434 g at birth and the other first twin baby girl weighed 1532 g at birth. The neonate remained hemodynamically stable with normal vital parameters. Anthropometric parameters at birth as per modified fenton's chart were weight 1434 g (3rd to 10th centile) length (50th-90th) and head circumference-32 cm. (50th to 90th).
The neonate was shifted to NICU was started on partial parenteral nutrition and enteral feeds with pasteurised donor human milk and the feeds were gradually hiked. He was initiated on kangaroo mother care and received other routine NICU care. Investigations including sepsis workup were normal. The feeds were gradually incremented by 30 ml/kg/day and full feeds were reached by day six of life. On day of life (DOL) 14 neonate developed late onset sepsis with pneumonia requiring non-invasive respiratory support, Inj.Piperacillin and Tazobactum (100 mg/kg/dose in 3 times a day, IV) and Inj.Amikacin (15 mg/kg/dose once a day, IV) was given for 10 days. Blood culture showed no growth.
Despite adequate feed volume and fortification of breast milk there was poor postnatal weight gain (average of 5-10 g/kg/day) and even by day 21 the neonate had not regained birth weight. Workup for sepsis including urinary tract infection, anaemia, osteopenia of prematurity and dyselectrolytemia was normal. By DOL 23 there was new onset clinical jaundice visible up to thighs and there was dark urine stain of diaper and stools were acholic. There was no fever and vital parameters were normal. Liver was 2.5 cm below right costal margin, non tender, soft in consistency. Left lobe was not palpable and there was no splenomegaly. Liver function tests done showed SGOT/SGPT-280/137, Total Bilirubin/Direct bilirubin-9.1 mg/dl/3.93mg/dl, albumin-2.5 mg/dl. INR- 1.2. Thyroid function test was normal. Repeat sepsis workup was negative. No previous liver function test was done. Neonate was diagnosed to have neonatal cholestasis and ultrasound abdomen showed fusiform dilatation noted including entire length of extrahepatic segment (1.2x0.5 cm) with distended gallbladder. In view of suspected choledochal cyst (CC), Magnetic resonance cholangiopancreatography (MRCP) was done which showed common bile duct (CBD) with fusiform dilatation in the proximal and mid portion with maximum diameter of 3.2 mm. Length of dilated segment was 1.3 cm. There was no distal obstruction with no filling defects with no intrahepatic bile duct dilatation (IHBRD). Right lobe of liver measured 5.6 cm, normal in size and signal intensity with normal contour and margins. Gall bladder was well distended. Pancreas appeared normal. Main pancreatic duct was not dilated with no evidence of anomalous pancreato-biliary duct union (APBDU). Spleen measured 4 cm, appeared normal in size, shape and signal intensity. There was no evidence of cholangitis. Based on clinical and radiological findings a diagnosis of Type 1c choledochal cyst was made. The antenatal scans were reviewed again and there was no choledochal cyst diagnosed prenatally. Since it was a twin pregnancy liver function tests and ultrasound of the abdomen of the other twin was done and was normal. The other twin had a normal NICU course with no jaundice, adequate weight gain and no complications.
Nutrition of the index twin was optimized to 160 kcal/kg/day, protein to 4 g/kg/day. He was started on preterm formula in addition to expressed breast milk via cup and spoon and direct breastfeeding. Neonate was also started on Vitamin A, Vitamin E, Vitamin D, Vitamin K and multivitamins. Oral cefixime prophylaxis was initiated along with oral ursodeoxycholic acid.
Paediatric surgery opinion was taken and early surgery has been planned upon adequate and sustained weight gain. Neonate was discharged on DOL-40 with weight of 1730 g. With adequate weight gain of 20-25 g/kg/day for 5 days. LFT before discharge showed SGOT/SGPT-280/137, Total Bilirubin/Direct bilirubin-5.3 mg/dl/2.9 mg/dl, albumin-2.5 mg/dl. Hearing screen, retinopathy of prematurity screen, ultrasound cranium and echocardiography were normal. Vaccination including hepatitis B were given as per age and neonate continues to be on multidisciplinary follow-up awaiting early surgery. Mother has been restarted on ribavirin and both the twins are currently 3 months old weighing 2800 g with normal growth and neurodvelopment, has been planned for surgery upon reaching weight of 3500 g and both twins will be evaluated for hepatitis C infection on follow-up.
| | | | Discussion | CC also known as congenital biliary dilatation are a group of rare congenital anomalies characterised by dilation of bile ducts. The incidence in Western population is reported to be 1 in 100000 – 150000 live births, although incidence as high as 1 in 1000, two third of reported cases have been reported from Japan1. The incidence of CC in our country is not known, though some studies have mentioned the anomaly to be not as uncommon as quoted in the Western studies.2 A female preponderance as high as 4:1 or 3:1 has been reported in the West.1
The most common variant of CC is Type 1(80-90% of all choledochal cyst).3 They are fusiform or spherical dilatation of extrahepatic biliary tree. The most popular hypothesis regarding the pathogenesis of choledochal cyst is APBDU which is defined by a union of pancreatic and biliary ducts outside the duodenal wall and proximal to ampulla of Vater. This explains the mechanism in 90% of cases. CC that develop in the absence of APBDU may be explained by other coexisting pathophysiological causes like a weak bile duct wall, distal biliary obstruction or sphincter of oddi dysfunction. Embryologic and motility disorders have also been postulated by some authors. An important implication to be noted here is that ABPDU associated choledochal cyst patients have more evidence of pathologically confirmed inflammation including hepatitis, cholangitis and pancreatitis. Some studies have noted various congenital anomalies like duodenal atresia, colonic atresia, gastroschisis, annular pancreas, pancreatic cyst and cardiac malformations to be associated with choledochal cysts. Type 1 and Type IV cysts are found to have the highest association with malignancy.4 Our index neonate had no evidence of APBDU on MRCP and had no other associated abnormality.
CC in neonates and infants universally presents with the typical jaundice associated with clay-coloured stools and high coloured urine similar to our patient’s presentation. Very rare cases in this group have also presented with duodenal obstruction with perforation, rupture of cyst and biliary peritonitis prompting emergency biliary drainage. Hepatic fibrosis is noted as a major complication in neonates. The incidence and grade of hepatic fibrosis is found higher in antenatally diagnosed neonates with CC who present early compared to infants who present after 1 month. Cirrhosis has been documented to be as high as 67% in these patients. In such early cases the histological changes observed are similar to the findings in biliary atresia.3
The primary diagnostic modality for choledochal cysts is a ultrasound, supported by other imaging techniques like computed tomography (CT), Magnetic Resonance Imaging (MRI),MRCP and to a lesser extent Percutaneous Transhepatic Cholangiography(PTC) and Endoscopic Retrograde Cholangiography(ERCP) due their invasiveness and associated risks. The higher imaging modalities are usually used to demarcate the extent of involvement of duct or extra hepatic tissue once a primary diagnosis of cyst is made in ultrasonography. ERCP and MRCP modalities are commonly found to be used in complicated cases for urgent management of cholangitis or obstructing biliary stones for stabilisation before definitive surgical management. Out of all the techniques mentioned above, MRCP facilitates better delineation of the biliary cyst subtype, pancreato-biliary ductal anatomy and further surgical planning.
Figure 1. Magnetic resonance cholangiopancreatography (MRCP) axial (top panel) and transverse section (bottom panel) showing common bile duct with fusiform dilatation in the proximal and mid portion with a maximum diameter of 3.2 mm.
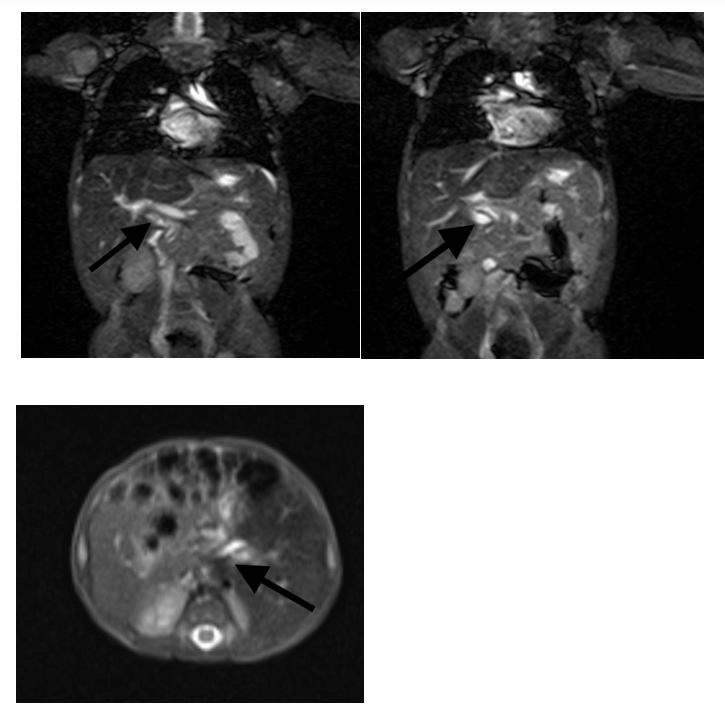
Cystic biliary atresia (CBA) patients which is a close differential to CC typically present earlier (<3 months of age) and their cysts appear smaller with less dilatation of the intrahepatic biliary system. An atretic gallbladder with irregular and hypoplastic biliary radicles is typical of CBA on ultrasound and cholangiography Conversely, infantile CC demonstrates a communication of the cyst with a dilated gallbladder in addition to a dilated intrahepatic biliary tree.
Cyst excision is the definitive management for CC. Historically different internal drainage procedures have been attempted, but have met with little success due to the persistence of biliary stasis. Complete surgical removal of all cystic tissue is advised as it is associated with a very high risk of malignancy.5 Reported incidences of malignancy in unoperated cases of choledochal cysts range from 10-15%.1 The surgical approach will be dependent on the specific type and subtype of the cyst, but generally aims to excise the cyst completely and restoration of biliary enteric drainage into duodenum or via Roux-en-Y hepaticojejunostomy.6
Even in developed countries where a larger chunk of these cases is antenatally diagnosed, the surgeries get postponed to 3-6 months owing to technical difficulties and associated anaesthetic risks during the neonatal period6. In antenatally diagnosed symptomatic cases of CC, many authors suggest an ideal time for definitive surgery to be anywhere between 2 weeks to 3 months, since liver damage is noted to develop early in these patients. Some neonates, irrespective of their antenatal diagnosis may be surprisingly asymptomatic: borderline elevation of bilirubin levels, gradually increasing size of the cyst and functional maturation of liver with advancement of age are some of the reasons which might explain asymptomatic status. Even in asymptomatic patients, many authors advocate early intervention, as delayed intervention is found to be associated with higher incidences of hepatic fibrosis, portal fibrosis, CBD strictures and cirrhosis.6 To conclude, all neonatal cases of choledochal cyst need early surgical intervention irrespective of their antenatal diagnosis or their clinically asymptomatic status so as to prevent liver damage, associated complications and to facilitate complete functional recovery.
Another complicated aspect in the management of this patient was the perinatal exposure of HCV which placed the infant at a very high risk of acquiring infection. The risk of transmission is as high as one to five percent, which is increased in presence of HIV co-infection.7,8,9 Though universal screening is not warranted in our country, current recommendations advise early testing in high-risk cases in a two-step process. Currently, there is no evidence in the literature regarding the direct causative role of perinatal hepatitis C infection in abnormal embryogenesis or immunological changes leading to the formation of choledochal cysts. The infant has to be tested for HCV RNA, between two to six months.10,11,12 A negative test may suggest a lower likelihood of HCV infection in the infant, while a positive test indicates the presence of infection. This does not predict chronicity, as there is a very high chance of spontaneous viral clearance.8 Irrespective of the result of the first test, all perinatally exposed infants need to be followed up at 18 months with anti- HCV antibody test. Positive tests warrant HCV RNA testing. Though treatment is deferred until 3 years in this population, these patients need regular monitoring of liver function tests for the progression of liver disease.8 | | | | Conclusion | | Choledochal cyst type 1c is to be considered in the differential of obstructive jaundice in a preterm neonate and requires early diagnosis with MRCP for better delineation of the biliary cyst subtype, pancreatic-biliary ductal anatomy and further surgical planning. It is imperative to diagnose perinatal HCV infection in those with maternal hepatitis C infection. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Singham J, Yoshida EM, Scudamore CH. Choledochal cysts: part 1 of 3: classification and pathogenesis. Can J Surg. 2009 Oct;52(5):434-40. PMID: 19865581; PMCID: PMC2769090.
- Poddar U, Thapa B.R, Chhabra M. Choledochal cysts in infants and children. Indian Pediatrics 1998; 35:613-18.
- Soares KC, Goldstein SD, Ghaseb MA, Kamel I, Hackam DJ, Pawlik TM. Pediatric choledochal cysts: diagnosis and current management. Pediatr Surg Int. 2017 Jun;33(6):637-650. doi: 10.1007/s00383-017-4083-6. Epub 2017 Mar 31. PMID: 28364277. [CrossRef]
- Soares KC, Arnaoutakis DJ, Kamel I, Rastegar N, Anders R, Maithel S, Pawlik TM. Choledochal cysts: presentation, clinical differentiation, and management. J Am Coll Surg. 2014 Dec;219(6):1167-80. doi: 10.1016/j.jamcollsurg.2014.04.023. Epub 2014 Jun 27. PMID: 25442379; PMCID: PMC4332770. [CrossRef]
- Jabłońska B. Biliary cysts: etiology, diagnosis and management. World J Gastroenterol. 2012 Sep 21;18(35):4801-10. doi: 10.3748/wjg.v18.i35.4801. PMID: 23002354. [CrossRef]
- Diao M, Li L, Cheng W. Timing of surgery for prenatally diagnosed asymptomatic choledochal cysts: a prospective randomized study. J Pediatr Surg. 2012 Mar;47(3):506-12. doi: 10.1016/j.jpedsurg.2011.09.056. PMID: 22424346. [CrossRef]
- Benova L, Mohamoud YA, Calvert C, Abu-Raddad LJ. Vertical transmission of hepatitis C virus: systematic review and meta-analysis. Clin Infect Dis. 2014 Sep 15;59(6):765-73. doi:10.1093/cid/ciu447. Epub 2014 Jun 13. PMID: 24928290; PMCID: PMC4144266. [CrossRef]
- AASLD/IDSA. HCV in Children: Recommendations for HCV Testing of Perinatally Exposed Children and Siblings of HCV-Infected Children. Available at: https://www.hcvguidelines.org/unique-populations/children
- Yeung LT, King SM, Roberts EA. Mother-to-infant transmission of hepatitis C virus.Hepatology. 2001 Aug;34(2):223-9. doi: 10.1053/jhep.2001.25885. PMID: 11481604. [CrossRef]
- Gowda C, Smith S, Crim L, Moyer K, Sánchez PJ, Honegger JR. Nucleic Acid Testing for Diagnosis of Perinatally Acquired Hepatitis C Virus Infection in Early Infancy. Clin Infect Dis. 2021 Nov 2;73(9):e3340-e3346. doi: 10.1093/cid/ciaa949. PMID: 32640018; PMCID:PMC8563185. [CrossRef]
- Ghany MG, Morgan TR; AASLD-IDSA Hepatitis C Guidance Panel. Hepatitis C Guidance 2019 Update: American Association for the Study of Liver Diseases-Infectious Diseases Society of America Recommendations for Testing, Managing, and Treating Hepatitis C Virus Infection. Hepatology. 2020 Feb;71(2):686-721. doi: 10.1002/hep.31060. PMID: 31816111; PMCID: PMC9710295. [CrossRef]
- Leung DH, Squires JE, Jhaveri R, Kerkar N, Lin CH, Mohan P, Murray KF, Gonzalez-Peralta RP, Roberts EA, Sundaram SS. Hepatitis C in 2020: A North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition Position Paper. J Pediatr Gastroenterol Nutr.2020 Sep;71(3):407-417. doi: 10.1097/MPG.0000000000002814. PMID: 32826718. [CrossRef]
DOI: https://doi.org/10.7199/ped.oncall.2026.30
|
| Cite this article as: | | RR P, Goyal M, Sreekumari J, Haribalakrishna A. Type 1c Choledochal cyst in a premature twin neonate: Case report and literature review. Pediatr Oncall J. 2024 May 22. doi: 10.7199/ped.oncall.2026.30 |
|