Giorgina Mieli-Vergani, Sarah Ann Tizzard.
Paediatric Liver,GI & Nutrition Centre, King's College London School of Medicine at King's College Hospital, London, UK.
ADDRESS FOR CORRESPONDENCE
Giorgina Mieli-Vergani, Professor of Paediatric Hepatology, Paediatric Liver, GI & Nutrition Centre, King's College Hospital, Denmark Hill, London SE59RS, UK.
Email: giorgina.vergani@kcl.ac.uk | INTRODUCTION
Biliary atresia (BA), a condition unique to infancy, is the end result of a destructive, idiopathic, inflammatory process affecting both intra and extra hepatic bile ducts, leading to fibrosis and obliteration of the biliary tract and to biliary cirrhosis. Cirrhosis and portal hypertension may appear as early as at 2 months of age, and without appropriate treatment most children die within the first 2 years of life. (1) BA is the most frequent surgically correctable liver disorder in infancy, as well as the most frequent indication for liver transplantation in paediatric age. Two forms of BA are described: a more common (90% of cases) peri or post natal isolated form, possibly virus related; and a less common (10% of cases) foetal or embryonic form, with a high frequency of associated malformations, such as BA splenic malformation syndrome (BASM), characterised by cardiovascular defects, polysplenia or asplenia, abdominal situs inversus, intestinal malrotation and positional abnormalities of the portal vein and hepatic artery. The cause of BA remains unknown.
EPIDEMIOLOGY AND PATHOGENESIS
BA is more common in Asia and the Pacific region than in the rest of the world. The reported incidence of BA varies between 1/3000 in French Polynesia to 1/20,000 in Holland and France. (2-9) Females are affected slightly more frequently than males. Some studies have suggested seasonal variation and clustering of BA cases (5-7), but this has not been confirmed in larger studies. (2,3,10,11).
Studies of bile duct remnants removed at surgery and from serial sectioning and reconstruction of surgical and post mortem liver specimens indicate that BA in most cases arises from a sclerosing inflammatory process affecting previously formed bile ducts. (12) The cause of such inflammatory process remains elusive. It is conceivable that BA represents a common final phenotypic pathway of neonatal liver injury caused by diverse causes, including developmental, vascular or infectious factors, which may act antenatally or within the first three months of life, in a genetically predisposed individual. (13,14) Though BA is not an inherited disorder, since identical twins are usually discordant for the disease, and occurrence of BA within the same family is exceedingly rare, (15,16) it is possible that a genetic predisposition to an aberrant immune response against an exogenous agent or somatic mutations of genes regulating bile duct morphogenesis in foetal life are involved. (14) Whatever the initiating event, as bile flow increases perinatally, bile leakage from the abnormal ducts is likely to trigger an intense inflammatory reaction, with consequent obliteration of the biliary tree. Bile extravasated into periductal tissues would lead to protracted inflammation and fibrosis, causing secondary obliteration and obstruction of the more distal bile ducts. The detergent effect of the extravasated bile, however, cannot be the only explanation for liver damage, since the disease can progress also in those patients in whom the Kasai portoenterostomy surgical procedure has achieved adequate bile flow. Proposed etiological factors in BA include defective morphogenesis/genetic factors, vascular abnormalities, viral infection, exposure to toxins and immune mechanisms. (14)
CLINICAL FEATURES AND DIAGNOSIS
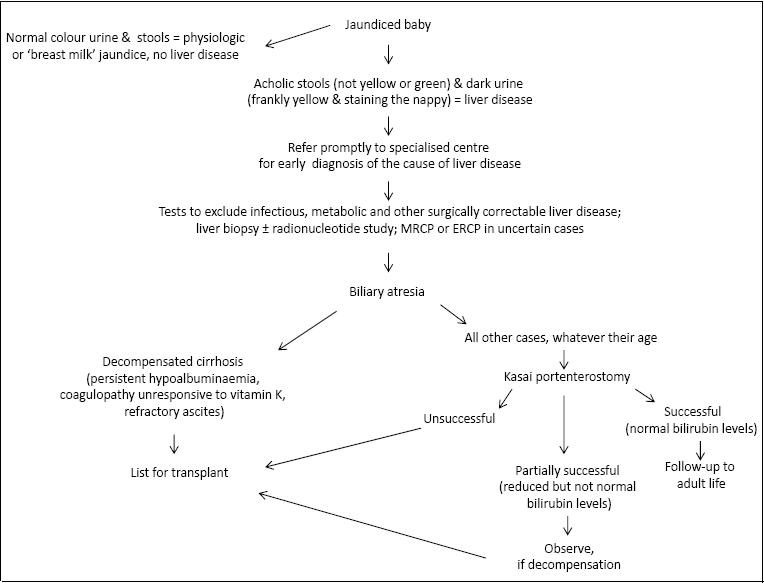
Clinical features of BA are jaundice, pale stools and dark urine presenting at or soon after birth. Diagnosis is unfortunately often delayed, because physiological jaundice, which at variance to jaundice due to liver disease is characterised by unconjugated bilirubinemia, is common in babies, and because infants with BA have no major symptoms apart from jaundice in the first few weeks of life, when corrective surgery would be most effective. This is a particular problem for infants with the perinatal/acquired form of BA, whose stools may have some pigment in the first few weeks of life, before bile flow is completely obstructed. (12,17) The presence of initially pigmented stools not uncommonly leads inexperienced health professionals to reassure parents that complete bile duct obstruction is unlikely. All jaundiced infants with pale or acholic stools (i.e. stools that are not green or yellow) at whatever age should be referred promptly to specialized centres because early surgical treatment of BA is essential for a good outcome, while late surgery carries a severe prognosis, particularly in BASM syndrome. (18-20) Hence, it is of paramount importance for health professionals attending infants to check the colour of the urine and stools of all jaundiced babies, irrespective of their general health or age, and refer promptly those with dark urine and pale stools to the appropriate centres. (3,4,18)
No satisfactory screening test for BA is available, though promising results using stool colour cards have been reported from Taiwan. (21) Physical examination and laboratory tests give little clue to the diagnosis of BA. Most of the affected infants have a mild degree of hepatomegaly and splenomegaly. Ascites or cutaneous signs of chronic liver disease are rarely detected in the early stages of the disease, when diagnosis is most important for effective surgical intervention. Biochemical findings are non specific, with levels of transaminases, gamma-glutamyl-transpeptidase and alkaline phosphatase similar to those found in other forms of neonatal cholestasis. Coagulopathy, if present, is responsive to intravenous vitamin K. An ultrasound scan revealing an absent or abnormal gallbladder with an irregular wall, (22) or, in older infants, the triangular cord sign (23), is suggestive of BA. However, a normal gallbladder or absence of the triangular cord sign, do not exclude BA. Histological examination of the liver by an experienced histopathologist leads to the correct diagnosis of BA in up to 90% of cases. (24) Typical histological findings are edematous portal tracts with inflammatory changes, bile duct proliferation and bile plugs, but in very young babies these features can be much less obvious than in older infants. A scan with 99 mTc-tagged iminodiacetic acid derivatives, such as methyl-brom-IDA, which have good hepatic uptake and relatively poor renal uptake, is often used in prolonged neonatal cholestasis in an attempt to differentiate BA from other forms of liver disease. Discrimination between BA and intrahepatic cholestasis is enhanced if the infant is pre-treated with phenobarbitone (5 mg/kg/day for at least 3 days). Repeated imaging up to 24 hours after intravenous injection may be required to establish whether the isotope has reached the gut. Radionucleide studies, however, are only useful if the isotope is demonstrated in the gut, thereby excluding BA and avoiding an unnecessary and potentially dangerous laparotomy, but absence of excretion in the gut does not equate with biliary atresia. Daily observation of the stool colour and trends in serum bilirubin levels are essential even if patency of the bile ducts is demonstrated. If the stools remain acholic, the liver biopsy should be repeated, and further investigations are necessary to exclude late-onset biliary atresia. These include nuclear magnetic resonance (25,26) or endoscopic retrograde cholangiography (27-29) to assess the patency of the biliary system. If they are not informative, an explorative laparotomy with intraoperative cholangiography is required, but should be undertaken by an experienced surgeon, since hypoplastic extrahepatic ducts caused by a severe intrahepatic cholestasis may be interpreted as atretic, leading to an unnecessary and possibly damaging operation. (30) The diagnosis of BA should be always confirmed by histological examination of the excised biliary remnants.
MANAGEMENT AND PROGNOSIS
The treatment of BA is surgical and aims at re-establishing bile flow. There are three macroscopic forms of BA: type I, affecting the distal part of the common duct; type II affecting the common hepatic duct, but sparing the gallbladder and common bile duct; and type III affecting right and left hepatic ducts and the gallbladder. The most common form is type III (85-90% cases), often referred to as 'uncorrectable', where the surgical reconstruction (porto-enterostomy) is most challenging. For these patients, the fibrous tissue at the porta hepatis is transected flush with the liver and a Roux-en-Y loop of jejunum is anastomosed around the fibrous edges of the transected tissue, forming a porto-enterostomy (Kasai procedure). (31,32) In the 5-10% of infants who have a patent common bile duct containing bile in continuity with intrahepatic bile ducts, a biliary-intestinal anastomosis via a Roux-en-Y loop of jejunum usually allows satisfactory bile drainage. (33)
The best preoperative predictor of a successful portoenterostomy is the age at surgery, emphasising the importance of early referral to a specialized centre. If the portoenterostomy is performed by an experienced surgeon good bile flow with normal serum bilirubin values can be achieved in more than 80% of children operated on by 60 days of age, but in only 20-30% with later surgery. (3,18,34,35) Data from Japan show an advantage for infants operated on when younger than 30 days of age, no difference in outcome in those operated on between 30 and 90 days, and a significant disadvantage only for those operated on later than 90 days. (36)
The success of the operation is judged by the appearance of pigment in the stools and clearance of jaundice. Reports from the UK show that the ultimate outcome of BA is improved by centralisation of care in specialised paediatric hepatology centres with expertise in both Kasai portoenterostomy and pediatric liver transplantation. (4,37) The importance of centralization of expertise for the successful management of BA has been confirmed in France. (38) If bilirubin returns to normal, a 90% 15-year survival has been reported (39), with a good quality of life in those who have reached the fourth decade. (40) If the bilirubin is not reduced by surgery, the rate of progression of cirrhosis is not slowed and survival beyond the second birthday is unusual. If partial bile drainage is obtained, development of end stage chronic liver disease may be delayed until puberty or early adult life. Eventually, 70-80% of patients undergoing Kasai portoenterostomy, even if surgery is successful, will require liver transplantation or succumb to their disease due to progressive liver failure. (41) At the other hand of the spectrum, 11% of children with surgically corrected BA are free of clinical and biochemical signs of liver disease after ten-year follow up. (42)
A serious postoperative complication is cholangitis, which occurs in over 50% of patients during the first two years after surgery, (43) and is due to a wide range of microorganisms. It is characterised by fever, recurrence or aggravation of jaundice, and frequently features of septicemia. Blood culture, ascitic aspirate or liver biopsy to identify the organism responsible should precede intravenous antibiotic therapy, which is continued for 14 days if a pathogen is identified. Often, however, the diagnosis of cholangitis is not obvious and unexplained fever may be the only symptom. Antibiotics are then started empirically, after taking a blood culture and assessing liver function tests, C-reactive protein and full blood count. If the fever responds to antibiotics, these are continued for 5 days. Should the fever recur after stopping antibiotics, a liver biopsy is performed for histological examination and culture. Long-term prophylaxis with rotating antibiotics may be indicated for recurrent cholangitis. (34)
In an attempt to improve bile flow and postoperative cholangitis rate, corticosteroids post surgery have been advocated in some centres. (44) Reasons for their use are their ability to increase bile flow by inducing canalicular electrolyte transport (45,46) and their immunological and anti-inflammatory effects. (47) There is, however, no solid clinical evidence that steroids are of benefit. Most published studies are retrospective and uncontrolled. (48-51) There are only two published prospective studies. Petersen et al (52) used a high-dose steroid regimen to treat 20 consecutive patients after Kasai portoenterostomy and compared them with a historical control group, while Davenport et al (53) performed a prospective, double-blinded, randomized, placebo-controlled trial of low dose oral prednisolone in 71 children with BA. Neither study showed any difference in overall survival, liver transplant frequency, survival with native liver, or jaundice-free survival with native liver.
Another complication, particularly in patients with BASM, is progressive hepatopulmonary syndrome, which at times becomes the primary indication for liver transplantation, as the hepatopulmonary symptoms slowly disappear after successful grafting.
A degree of portal hypertension is present in almost all cases of BA at the time of portoenterostomy. Approximately 50% of all survivors aged 5 years, even those with normal bilirubin levels, have oesophageal varices, but only 10-15% have gastrointestinal bleeding, which needs to be treated with banding or injection sclerotherapy. (34)
LIVER TRANSPLANTATION
Liver transplantation in BA remains complementary to portoenterostomy, except for infants who, because of delayed diagnosis, present with decompensated cirrhosis, i.e. with severely impaired liver synthetic function (persistent hypoalbuminemia, coagulopathy unresponsive to vitamin K, refractory ascites). For all other children, whatever the age at diagnosis, the Kasai procedure should be performed by an experienced surgeon, as in specialised centres good outcomes have been reported also in children operated on late. (19) Moreover, even if the portoenterostomy is only partially successful, it will allow the child to grow to a size more suitable for transplantation. A completely unsuccessful Kasai procedure, on the other hand, does not preclude early transplantation. The combination of Kasai portoenterostomy, followed by transplantation if complications arise, has considerably improved the survival of children with BA, with a 4-year actuarial survival with native liver of 51% and an overall (with native liver and post liver transplant) 4-year actuarial survival of 89%. (37)
Liver transplantation is a standard therapeutic option for children with an unsuccessful Kasai portoenterostomy, or who develop progressive liver disease or hepatopulmonary syndrome despite successful surgery. Liver transplantation, however, remains a formidable procedure. The recipient is likely to have one or more life-threatening complications in the perioperative or postoperative period. Lifelong immunosuppressive therapy is required, with a high risk of opportunistic and community-acquired infections, Epstein Barr virus associated lymphoproliferative disease, renal impairment, etc, necessitating close medical and surgical supervision. (54) Most of the survivors have a good quality of life and attend school, but significant learning issues have been reported in a considerable proportion of 823 paediatric liver transplant recipients, 34% requiring special education and 20% having to repeat grades. (55) The long-term medical and psychological effects of liver transplantation in childhood are as yet unknown.
The supply of donors of suitable size and blood group, even after the introduction of liver reduction techniques and with an increased use of split grafts, where one donor liver is used for two recipients - usually one child and one adult - remains a major limiting factor in liver transplantation. Hence, segmental graft transplant from living relatives is currently frequently used, with survival rates in excess of 90% in infants in whom Kasai portoenterostomy had been unsuccessful. (54,56,57) This procedure, however, introduces ethical considerations and morbidity and mortality risks for the donor, who must to be very carefully assessed before donation. (54, 58)
ALGORITHM FOR MANAGEMENT OF BILIARY ATRESIA (Fig 1)
PRACTICE POINTS
Biliary atresia is a disease unique to infancy, in which bile duct damage can initiate before or after birth: hence pigmented stools after birth do not exclude it
Biliary atresia is the most common cause of severe liver disease in infancy, but at the time surgery is most effective (before 60 days of age) babies usually have no signs of ill health apart from persistent jaundice
Jaundice in babies is often ignored because unconjugated hyperbilirubinemia (physiological jaundice, with normal colour of urine and stools) is common in neonates
Biliary atresia should be suspected in all infants with acholic stools (i.e. not green or yellow) and pigmented urine (i.e. frankly yellow and staining the nappy), which indicate conjugated hyperbilirubinemia (i.e. liver disease), whatever their age and general health
Infants with suspected liver disease should be referred promptly to specialised paediatric hepatology centres, as early diagnosis and surgical intervention in biliary atresia is life saving
Cholestatic babies must be treated promptly with fat-soluble vitamins (A, D, E and K) to avoid serious complications, particularly intracranial bleeding caused by vitamin K deficiency
Kasai portoenterostomy followed by liver transplantation in those patients in whom the Kasai procedure is unsuccessful results in survival rates approaching 90%
Primary liver transplantation should be offered only to infants with decompensated cirrhosis at the time of diagnosis, i.e. those with persistent hypoalbuminemia, coagulopathy unresponsive to vitamin K, and refractory ascites | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Samyn M, Mieli-Vergani G. Liver and biliary disease in infancy. Medicine 2011;39:565-570. [CrossRef]
- Houwen RH, Kerremans, II, van Steensel-Moll HA, van Romunde LK, Bijleveld CM, Schweizer P. Time-space distribution of extrahepatic biliary atresia in The Netherlands and West Germany. Z Kinderchir 1988;43(2):68-71. [CrossRef]
- Chardot C, Carton M, Spire-Bendelac N, Le Pommelet C, Golmard JL, Auvert B. Epidemiology of biliary atresia in France: a national study 1986-96. J Hepatol 1999;31:1006-1013. [CrossRef]
- McKiernan PJ, Baker AJ, Kelly DA. The frequency and outcome of biliary atresia in the UK and Ireland. Lancet 2000;355:25-29. [CrossRef]
- Strickland AD, Shannon K. Studies in the etiology of extrahepatic biliary atresia: time-space clustering. J Pediatr 1982;100:749-753. [CrossRef]
- Danks DM, Campbell PE, Jack I, Rogers J, Smith AL. Studies of the aetiology of neonatal hepatitis and biliary atresia. Arch Dis Child 1977;52:360-367. [CrossRef]
- Yoon PW, Bresee JS, Olney RS, James LM, Khoury MJ. Epidemiology of biliary atresia: a population-based study. Pediatrics 1997;99:376-382. [CrossRef]
- Shim WK, Kasai M, Spence MA. Racial influence on the incidence of biliary atresia. Prog Pediatr Surg 1974;6:53-62. [PubMed]
- Vic P, Gestas P, Mallet EC, Arnaud JP. Biliary atresia in French Polynesia. Retrospective study of 10 years. Arch Pediatr 1994;1:646-651. [PubMed]
- Ayas MF, Hillemeier AC, Olson AD. Lack of evidence for seasonal variation in extrahepatic biliary atresia during infancy. J Clin Gastroenterol 1996;22:292-294. [CrossRef]
- Davenport M, Dhawan A. Epidemiologic study of infants with biliary atresia. Pediatrics 1998;101:729-730. [CrossRef]
- Gautier M, Eliot N. Extrahepatic biliary atresia. Morphological study of 98 biliary remnants. Arch Pathol Lab Med 1981;105:397-402. [PubMed]
- Sokol RJ, Mack C. Etiopathogenesis of biliary atresia. Sem Liver Dis 2001;21:517-524. [CrossRef]
- Mieli-Vergani G, Vergani D. Biliary atresia. Semin Immunopathol 2009;31:371-381. [CrossRef]
- Schweizer P, Kerremans J. Discordant findings in extrahepatic bile duct atresia in 6 sets of twins. Z Kinderchir 1988;43:72-75. [CrossRef]
- Hyams JS, Glaser JH, Leichtner AM, Morecki R. Discordance for biliary atresia in 2 sets of monozygotic twins. Journal of Pediatrics 1985;107:420-422. [CrossRef]
- Desmet VJ. Congenital diseases of intrahepatic bile ducts: variations on the theme "ductal plate malformation". Hepatology 1992;16:1069-1083. [CrossRef]
- Mieli-Vergani G, Howard ER, Portman B, Mowat AP. Late referral for biliary atresia: missed opportunities for effective surgery. Lancet 1989;1:421-423. [CrossRef]
- Davenport M, Puricelli V, Farrant P, et al. The outcome of the older (>/=100 days) infant with biliary atresia. J Pediatr Surg 2004;39:575-581. [CrossRef]
- Davenport M, Caponcelli E, Livesey E, Hadzic N, Howard E. Surgical outcome in biliary atresia: etiology affects the influence of age at surgery. Ann Surg 2008;247:694-8. [CrossRef]
- Chen SM, Chang MH, Du JC, et al. Screening for biliary atresia by infant stool color card in Taiwan. Pediatrics 2006;117:1147-1154. [CrossRef]
- Farrant P, Meire HB, Mieli-Vergani G. Ultrasound features of the gall bladder in infants presenting with conjugated hyperbilirubinaemia. Br J Radiol 2000;73:1154-1158. [CrossRef]
- Park WH, Choi SO, Lee HJ, Kim SP, Zeon SK, Lee SL. A new diagnostic approach to biliary atresia with emphasis on the ultrasonographic triangular cord sign: comparison of ultrasonography, hepatobiliary scintigraphy, and liver needle biopsy in the evaluation of infantile cholestasis. J Pediatr Surg 1997;32:1555-1559 [CrossRef]
- Lai MW, Chang MH, Hsu SC, et al. Differential diagnosis of extrahepatic biliary atresia from neonatal hepatitis: a prospective study. J Pediatr Gastroenterol Nutr. 1994;18:121-127. [CrossRef]
- Guibaud L, Lachaud A, Touraine R, et al. MR cholangiography in neonates and infants: feasibility and preliminary applications. Am J Roentgenol 1998;170:27-31. [CrossRef]
- Norton KI, Glass RB, Kogan D, Lee JS, Emre S, Shneider BL. MR cholangiography in the evaluation of neonatal cholestasis: Initial results. Radiology 2002;222:687-692. [CrossRef]
- Wilkinson ML, Mieli-Vergani G, Ball C, Portmann B, Mowat AP. Endoscopic retrograde cholangiopancreatography in infantile cholestasis. Arch Dis Child 1991;66:121-123. [CrossRef]
- Iinuma Y, Narisawa R, Iwafuchi M, et al. The role of endoscopic retrograde cholangiopancreatography in infants with cholestasis. J Pediatr Surg 2000;35:545-549. [CrossRef]
- Shanmugam N, Harrison PM, Devlin J, et al. Selective use of endoscopic retrograde cholangiopancreatography in diagnosis of biliary atresia in infants younger than 100 days. J Pediatr Gastroenterol Nutr 2009;49:435-41. [CrossRef]
- Markowitz J, Daum F, Kahn EI, et al. Arteriohepatic dysplasia. I. Pitfalls in diagnosis and management. Hepatology 1983;3:74-76. [CrossRef]
- Kasai M, Suzuki S. A new operation for "non-correctable" biliary atresia: Hepatic porto-enterostomy. Shuiyutsu 1959;13:733-739.
- Ohi R. Surgery for biliary atresia. Liver 2001;21:175-182. [CrossRef]
- McClement JW, Howard ER, Mowat AP. Results of surgical treatment for extrahepatic biliary atresia in United Kingdom 1980-2. Survey conducted on behalf of the British Paediatric Association Gastroenterology Group and the British Association of Paediatric Surgeons. Br Med J 1985;290:345-347. [CrossRef]
- Davenport M, Kerkar N, Mieli-Vergani G, Mowat AP, Howard ER. Biliary atresia: the King's College Hospital experience (1974-1995). J Pediatr Surg 1997;32:479-485. [CrossRef]
- Ohi R. Biliary atresia. A surgical perspective. Clin Liver Dis 2000;4:779-804. [CrossRef]
- Nio M, Ohi R, Miyano T, Saeki M, Shiraki K, Tanaka K. Five- and 10-year survival rates after surgery for biliary atresia: A report from the Japanese Biliary Atresia Registry. J Pediatr Surg 2003;38:997-1000. [CrossRef]
- Davenport M, De Ville de Goyet J, Stringer MD, et al. Seamless management of biliary atresia in England and Wales (1999-2002). Lancet 2004;363:1354-1357. [CrossRef]
- Serinet MO, Broue P, Jacquemin E, et al. Management of patients with biliary atresia in France: results of a decentralized policy 1986-2002. Hepatology 2006;44:75-84. [CrossRef]
- Ohi R, Nio M, Chiba T, Endo N, Goto M, Ibrahim M. Long-term follow-up after surgery for patients with biliary atresia. J Pediatr Surg 1990;25:442-445. [CrossRef]
- Chiba T, Ohi R, Nio M, Ibrahim M. Late complications in long-term survivors of biliary atresia. Eur J Pediatr Surg 1992;2:22-25. [CrossRef]
- Davenport M. Biliary atresia. Semin Pediatr Surg 2005;14:42-48. [CrossRef]
- Hadzic N, Davenport M, Tizzard S, Singer J, Howard ER, Mieli-Vergani G. Long-term survival following Kasai portoenterostomy: is chronic liver disease inevitable? J Pediatr Gastroenterol Nutr 2003;37:430-433. [CrossRef]
- Ohkohchi N, Chiba T, Ohi R, Mori S. Long-term follow-up study of patients with cholangitis after successful Kasai operation in biliary atresia: selection of recipients for liver transplantation. J Pediatr Gastroenterol Nutr 1989;9:416-420. [CrossRef]
- Karrer FM, Lilly JR. Corticosteroid therapy in biliary atresia. J Pediatr Surg 1985;20:693-695. [CrossRef]
- Alvaro D, Gigliozzi A, Marucci L, et al. Corticosteroids modulate the secretory processes of the rat intrahepatic biliary epithelium. Gastroenterology 2002;122:1058-1069. [CrossRef]
- Miner PB, Jr., Gaito JM. Bile flow in response to pharmacologic agents. Hepatic DNA as a reference standard. Biochem Pharmacol 1979;28:1063-1066. [CrossRef]
- Hsieh CS, Huang CC, Huang LT, Tsai YJ, Chou MH, Chuang JH. Glucocorticoid treatment down-regulates chemokine expression of bacterial cholangitis in cholestatic rats. J Pediatr Surg 2004;39:10-15. [CrossRef]
- Vejchapipat P, Passakonnirin R, Sookpotarom P, Chittmittrapap S, Poovorawan Y. High-dose steroids do not improve early outcome in biliary atresia. J Pediatr Surg 2007;42:2102-2105. [CrossRef]
- Escobar MA, Jay CL, Brooks RM, et al. Effect of corticosteroid therapy on outcomes in biliary atresia after Kasai portoenterostomy. J Pediatr Surg 2006;41:99-103. [CrossRef]
- Stringer MD, Davison SM, Rajwal SR, McClean P. Kasai portoenterostomy: 12-year experience with a novel adjuvant therapy regimen. J Pediatr Surg 2007;42:1324-1328. [CrossRef]
- Shimadera S, Iwai N, Deguchi E, Kimura O, Fumino S, Ono S. The significance of steroid therapy after hepatoportoenterostomy in infants with biliary atresia. Eur J Pediatr Surg 2007;17:100-103. [CrossRef]
- Petersen C, Harder D, Melter M, et al. Postoperative high-dose steroids do not improve mid-term survival with native liver in biliary atresia. Am J Gastroenterol 2008;103:712-719. [CrossRef]
- Davenport M, Stringer MD, Tizzard SA, McClean P, Mieli-Vergani G, Hadzic N. Randomized, double-blind, placebo-controlled trial of corticosteroids after Kasai portoenterostomy for biliary atresia. Hepatology. 2007;46:1821-1827. [CrossRef]
- Vilca-Melendez H, Mieli-Vergani G. Pediatric liver transplantation. In: ?Organ Transplantation ? A Clinical Guide?. AA Klein, CJ Lweis, JC Madsen Eds. Cambridge University Press, Cambridge, UK. 2011;220-230. [CrossRef]
- Gilmour SM, Sorensen LG, Anand R, Yin W, Alonso EM; SPLIT Research Consortium. Liver Transpl 2010;16:1041-1048. [CrossRef]
- Ozawa K, Uemoto S, Tanaka K et al. An appraisal of pediatric liver transplantation from living relatives. Initial clinical experience in 20 pediatric liver transplantations from living relatives as donors. Ann Surgery 1992;216:547-553. [CrossRef]
- Karnsakul W, Intihar P, Konewko R, et al. Living donor liver transplantation in children: A single North American center experience over two decades. Pediatr Transplant 2012;16:486-95. [CrossRef]
- Miller CM, Smith ML, Diago Uso T. Living donor liver transplantation: ethical considerations. Mt Sinai J Med. 2012;79:214-22. [CrossRef]
DOI: https://doi.org/10.7199/ped.oncall.2012.59
|
| Cite this article as: | | Mieli-Vergani G, Tizzard S A. BILIARY ATRESIA AND KASAI`S SURGERY - WHEN IS IT TOO LATE. Pediatr Oncall J. 2012;9: 60-65. doi: 10.7199/ped.oncall.2012.59 |
|