Rodrigo Liberal1, Diego Vergani1, Giorgina Mieli-Vergani2.
1Institute of Liver Studies, King’s College London, UK,
2Paediatric Liver,GI and Nutrition Centre, School of Medicine at King’s College Hospital, London, UK.
ADDRESS FOR CORRESPONDENCE
Giorgina Mieli-Vergani, Professor of Paediatric Hepatology, Paediatric Liver, GI & Nutrition Centre, King’s College Hospital, Denmark Hill, London SE59RS, UK.
Email: giorgina.vergani@kcl.ac.uk | | Abstract | | Autoimmune liver disorders in childhood include autoimmune hepatitis (AIH) and autoimmune sclerosing cholangitis (ASC). These inflammatory liver disorders are characterised histologically by interface hepatitis, biochemically by elevated transaminase levels and serologically by autoantibodies and increased levels of immunoglobulin G. AIH is particularly aggressive in children and progresses rapidly unless immunosuppressive treatment is started promptly. With appropriate treatment some 80% of patients achieve remission and long-term survival. For non-responders and difficult-to-treat patients, novel and more effective therapeutic approaches are sought. ASC responds to the same treatment used for autoimmune hepatitis in regards to parenchymal inflammation, but bile duct disease progresses in about 50% of cases, leading to a worse prognosis and a higher liver transplantation requirement; moreover, it has a high recurrence rate after transplant. Progression of liver disease and recurrence after transplant are more common in patients with associated poorly controlled inflammatory bowel disease, which therefore needs to be treated aggressively and effectively. | | | | Keywords | | autoimmune hepatitis, autoimmune sclerosing cholangitis, immunosuppressive drugs, liver transplant | | | | Introduction | In pediatrics, there are two liver disorders where liver damage most likely stems from an autoimmune attack: “classical” autoimmune hepatitis (AIH) and AIH-sclerosing cholangitis overlap syndrome.
The presentation of childhood autoimmune liver disease (AILD) is non-specific and mimics most other liver disorders. Since prompt treatment is life saving, it is important to suspect AILD and perform appropriate investigations in all children who present with cryptogenic liver disorders.
At the King´s College Hospital Pediatric Hepatology tertiary referral centre, there has been an increase in the yearly incidence of juvenile AILD, which can only be partially explained by referral bias: in the 1990s, the yearly incidence of children over 4 months of age referred with AILD accounted for 2.3% of about 400 new cases, whereas in the new millennium the yearly incidence has increased to 12%. (unpublished data)
Autoimmune hepatitis
AIH is a progressive inflammatory liver disorder characterized serologically by high transaminases and immunoglobulin G (IgG) levels, presence of autoantibodies, and a histological picture of interface hepatitis (i.e. dense inflammatory mononuclear/plasma cell infiltrate of the portal tracts that invades the parenchyma), in the absence of a known aetiology. (1) Compared to adult patients, children and adolescents more frequently present acutely, and follow a more aggressive course. AIH usually responds to immunosuppressive treatment, which should be instituted as soon as the diagnosis is made. If left untreated, AIH generally progresses to liver failure requiring transplantation. (1)
Two types of AIH are recognized: type 1 AIH (AIH-1), positive for anti-nuclear (ANA) and/or smooth muscle antibodies (SMA), and type 2 AIH (AIH-2), positive for liver kidney microsomal type 1 (anti-LKM-1) and/or anti-liver cytosol type 1 autoantibodies (anti-LC-1). (2,3) While AIH-1 affects both children and adults, AIH-2 is predominantly a pediatric condition. IgG is usually raised at presentation in both types, though 15% of children with AIH-1 and 25% of those with AIH-2 have normal levels. IgA deficiency is common in AIH-2. (4) Severity of disease is similar in the two types, but children with AIH-2 have higher levels of bilirubin and transaminases at presentation than those with AIH-1 and present significantly more frequently with fulminant hepatic failure. (4) Excluding children with the fulminant presentation, a severely impaired hepatic synthetic function (i.e. prolonged prothrombin time and hypoalbuminemia) is more common in AIH-1 than in AIH-2. The severity of interface hepatitis at diagnosis is similar in both types, but cirrhosis on initial biopsy is more frequent in AIH-1 than in AIH-2, suggesting a more chronic course of disease in the former. Progression to cirrhosis during treatment is more frequent in AIH-1.
In both AIH types, a more severe disease course and a higher tendency to relapse are associated with the possession of antibodies to soluble liver antigen (SLA), which are present in approximately half of the patients with either type of AIH at diagnosis. (5) In both types, about 20% of patients have associated autoimmune disorders—including thyroiditis, vitiligo, type 1 diabetes, inflammatory bowel disease, and nephrotic syndrome—and about 40% have a family history of autoimmune disease. (4)
In the pediatric setting, the presentation of the disease follows one of three patterns:
| 1) |
In 40% of patients, the presentation is indistinguishable from that
of an acute viral hepatitis, being associated with non-specific symptoms
of malaise, nausea/vomiting, anorexia, and abdominal pain, followed by
jaundice, dark urine, and pale stools. Some children, particularly those
who are anti-LKM-1-positive, develop acute liver failure with grade II
to IV hepatic encephalopathy within 2 to 8 weeks from the onset of
symptoms. |
| 2) |
In 25 to 40% of patients, the onset is
insidious, with an illness characterized by progressive fatigue,
relapsing jaundice, headache, anorexia, amenorrhea, and weight loss,
lasting for several months and even years before diagnosis. |
| 3) |
In 10% of patients, there is no history of
jaundice, and the diagnosis follows a presentation with complications of
portal hypertension, such as upper gastrointestinal bleeding or
hypersplenism. (6) |
The mode of presentation of AIH in childhood is therefore variable, and the disease should be suspected and excluded in all children presenting with symptoms and signs of liver disease not ascribable to more common pathologies. The course of the disease can be fluctuating, with flares and spontaneous remissions, a pattern that may result in delayed referral and diagnosis. The majority of children, however, on physical examination have clinical signs of an underlying chronic liver disease, including cutaneous stigmata, firm liver and splenomegaly. At ultrasound, the liver parenchyma is often nodular and heterogeneous.
Autoimmune sclerosing cholangitis
In pediatrics, sclerosing cholangitis is often associated with florid autoimmune features, including elevated autoantibody titres and IgG levels as well as interface hepatitis. (7) This AIH/sclerosing cholangitis overlap syndrome, called autoimmune sclerosing cholangitis (ASC) has the same prevalence as AIH-1 in childhood, as shown in a prospective study conducted over a period of 16 years. (7) In this study, all children with serological and histological features of AILD underwent a cholangiogram at the time of presentation. Approximately 50% of the patients had bile duct changes characteristic of sclerosing cholangitis and were diagnosed as having ASC. A quarter of the children with ASC, despite abnormal cholangiograms, had no histological features suggesting bile duct involvement, and the diagnosis of sclerosing cholangitis was only possible because of the cholangiographic studies. Virtually all ASC patients were seropositive for ANA and/or SMA. ASC was diagnosed in a similar proportion of boys and girls.
The mode of presentation of ASC was similar to that of AIH-1. Inflammatory bowel disease, mostly asymptomatic or paucisymptomatic, was present in 45% of children with ASC compared to 20% of those with typical AIH, and 90% of children with ASC had greatly increased serum IgG levels. At presentation, standard liver function tests did not help in discriminating between AIH and ASC. Atypical perinuclear anti-neutrophil cytoplasmic antibody was present in 74% of patients with ASC compared with 45% of patients with AIH-1 and 11% of those with AIH-2. Evolution from AIH to ASC was documented, suggesting that AIH and ASC may be part of the same pathogenic process. (7)
Diagnosis
Diagnosis of AIH is based on a series of positive and negative criteria developed by the International Autoimmune Hepatitis Group (IAIHG) for adult patients. (8,9) The IAIHG scoring system was devised mainly for research purposes to allow ready comparison between series from different centres, but has also been used clinically, including in pediatric series. (4,10) More recently the IAIHG have published a simplified scoring system based on autoantibodies, IgG, histology, and exclusion of viral hepatitis that is better suited to clinical application. (11) However, neither scoring system is readily applicable to juvenile AIH, where diagnostically relevant autoantibodies often have titres lower than the cut-off value considered positive in adults. Thus, in pediatrics immunofluorescence titres – as determined on rodent liver, kidney and stomach substrates – of 1:20 for ANA and SMA and of 1:10 for anti-LKM-1 are significant. In addition, neither system can distinguish between AIH and ASC (12), which can only be differentiated if a cholangiogram is performed at presentation (Figure 1).
Treatment
Standard treatment of autoimmune hepatitis
The conventional treatment of childhood AIH consists of predniso(lo)ne at 2 mg/kg/day (maximum 60 mg/day), which is gradually decreased over 4-8 weeks, in parallel to the declining transaminase levels, towards a maintenance dose of 2.5-5 mg/day, depending on age and weight, (13,14) Within the initial two months of treatment, an 80% decrease in serum aminotransferase levels is commonly achieved, but complete normalization can take several months. (4) During the first 6-8 weeks of treatment, liver function tests should be performed often to allow weekly dose adjustments while avoiding severe steroid side effects. (14) In our Centre, azathioprine is added as a steroid sparing agent when the transaminase levels stop decreasing on prednisolone alone or in the presence of early, serious steroid side effects (e.g. psychosis). azathioprine is used at a starting dose of 0.5 mg/kg/day, which, in the absence of signs of toxicity, is increased up to a maximum of 2.0–2.5 mg/kg/day until biochemical control of the disease is achieved. Centres differ in terms of the time at which azathioprine is utilised; in some it is added in all cases at a dose of 0.5-2 mg/kg/day after a few weeks of steroid treatment, and in others a combination of steroids and azathioprine is used from the outset. However, caution is recommended, particularly in severely jaundiced patients, given the hepatotoxic properties of azathioprine. Regardless of the initial choice of treatment protocol, 85% of patients eventually require the addition of azathioprine. (14) Relapses on treatment are seen in some 40% of patients, particularly adolescents, and are often due to non-adherence. (15) Treatment is usually for life. Only some 20% of patients with AIH-1 can stop immunosuppressive therapy successfully, while this is very rarely achieved in AIH-2. Children are usually treated for at least 3 years and careful suspension of treatment should be considered only if a follow-up liver biopsy shows no evidence of inflammation after at least one year of normal transaminase and IgG levels and negative or very low titre autoantibodies, tested at 3-monthly intervals.
Alternative treatment
Alternative treatment regimens have been proposed. Firstly, remission has been induced in treatment naïve children using cyclosporine A alone for six months, followed by addition of prednisone and azathioprine. cyclosporine is then discontinued one month later. (16) However, whether this protocol has any advantage over standard treatment has not yet been evaluated in controlled studies. Secondly, budesonide has been used in a large European study in combination with azathioprine. (17) budesonide is an attractive alternative to predniso(lo)ne, as it has a hepatic first-pass clearance of > 90% of oral dose and fewer side effects, although it cannot be used in cirrhotic patients, who represent a large proportion of AIH cases. However, in the pediatric cohort of this study, budesonide and azathiprine were disappointing compared to a combination of medium-dose prednisone and azathioprine; there were similarly low remission rates of 16% for budesonide and 15% for prednisone after 6 months of treatment and of 50% and 42% respectively after 12 months of treatment. (18) Hence, further studies are needed before budesonide can be recommened for the treatment of juvenile AIH. (19) Despite this, budesonide could be a valid alternative in selected non-cirrhotic patients at risk of adverse steroids side effects.
Treatment of refractory cases
Mycophenolate mofetil (MMF) - a prodrug of mycophenolic acid that affects purine synthesis - in association with prednisolone has been successfully used in those children resistant to standard immunosuppression or intolerant to azathioprine (up to 10%). (20) In the absence of response or intolerance for mmf (headache, diarrhea, nausea, dizziness, hair loss, and neutropenia), calcineurin inhibitors should be considered. In our centre, tacrolimus, in combination with prednisolone, has been successful in inducing remission in difficult-to-treat patients.
Treatment of autoimmune sclerosing cholangitis
ASC responds to the same immunosuppressive treatment described above for AIH if treatment is started early, with resolution of liver test abnormalities within a few months in most patients; however, its medium- to long-term prognosis is worse than that of AIH because of progression of bile duct disease despite treatment in 50% of patients. (7) Ursodeoxycholic acid (UDCA) is usually added to steroids and azathioprine, but whether it is helpful in arresting the progression of the bile duct disease remains to be established. In adults with primary sclerosing cholangitis high-dose udca was reported as more beneficial than standard doses (21), but a randomized double-blind controlled study from the Mayo Clinic shows that high-dose udca has a negative long-term effect. (22) It is prudent, therefore, to use doses not higher than 15-20 mg/kg/day. ASC is often associated with inflammatory bowel disease which should be investigated even in the absence of symptoms and appropriately treated. Flares of the liver disease often follow flares of the intestinal disease.
Liver transplantation
Approximately 10% of children with AIH and 20% of those with ASC require liver transplantation. Liver transplantation is indicated in patients who present with fulminant hepatic failure (i.e. with encephalopathy) and in those who develop end-stage liver disease despite treatment. After transplantation, recurrent AIH is reported in about 20% of cases (23) and recurrent ASC in about 70%. (24) Diagnosis of recurrence is based on biochemical abnormalities, seropositivity for autoantibodies, interface hepatitis on histology, steroid dependence, and, for ASC, presence of cholangiopathy. Recurrence of ASC after transplant is often associated to poorly controlled inflammatory bowel disease. Recurrence may even appear years after transplantation, therefore steroid-based immunosuppression should be maintained at a higher dose than that used for patients transplanted for non-autoimmune liver diseases. (24)
Figure 1. Algorithm for treatment decision in children with autoimmune liver disease. (Adapted from reference 6.)
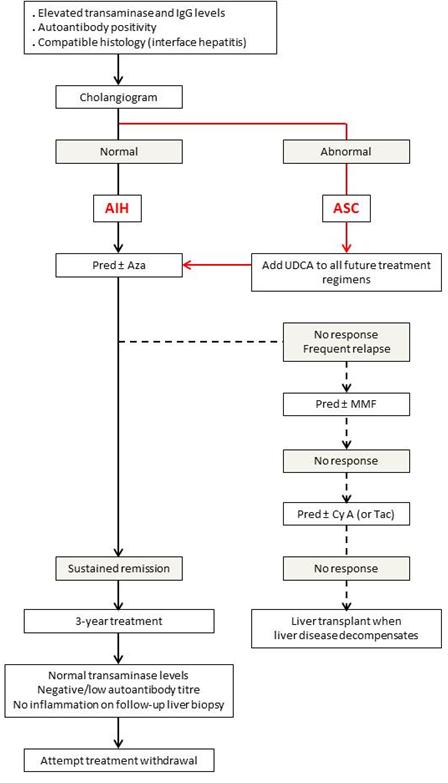
IgG: immunoglobulin G, AIH: autoimmune hepatitis, ASC: autoimmune sclerosing cholangitis, Pred: prednisolone, Aza: azathioprine, udca: ursodeoxycholic acid, mmf: mycophenolate mofetil, CyA: cyclosporine A, Tac: tacrolimus | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Mieli-Vergani G, Vergani D. Autoimmune hepatitis. Nat Rev Gastroenterol Hepatol 2011;8:320-329. [CrossRef]
- Vergani D, Choudhuri K, Bogdanos DP, Mieli-Vergani G. Pathogenesis of autoimmune hepatitis. Clin Liver Dis 2002;6:727-737. [CrossRef]
- Vergani D, Alvarez F, Bianchi FB, Cançado EL, Mackay IR, Manns MP, et al. Liver autoimmune serology: a consensus statement from the committee for autoimmune serology of the International Autoimmune Hepatitis Group. J Hepatol 2004;41:677-683 [CrossRef]
- Gregorio GV, Portmann B, Reid F, Doherty D, Donaldson P, McFarlane B, et al. Autoimmune hepatitis in childhood: a 20-year experience. Hepatology 1997;25:541-547. [CrossRef]
- Ma Y, Okamoto M, Thomas MG, Bogdanos DP, Lopes AR, Portmann B, et al. Antibodies to conformational epitopes of soluble liver antigen define a severe form of autoimmune liver disease. Hepatology 2002;35:658-664 [CrossRef]
- Mieli-Vergani G, Vergani D. Autoimmune liver diseases in children - what is different from adulthood? Best Pract Res Clin Gastroenterol 2011;25:783-795. [CrossRef]
- Gregorio GV, Portmann B, Karani J Harrison P, Donaldson PT, Vergani D, et al. Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: a 16-year prospective study. Hepatology 2001;33:544-553 [CrossRef]
- Johnson PJ, McFarlane IG. Meeting report: International Autoimmune Hepatitis Group. Hepatology 1993;18:998-1005. [CrossRef]
- Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, et al. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol 1999;31:929-938 [CrossRef]
- Ebbeson RL, Schreiber RA. Diagnosing autoimmune hepatitis in children: is the International Autoimmune Hepatitis Group scoring system useful? Clin Gastroenterol Hepatol 2004;2:935-940. [CrossRef]
- Hennes EM, Zeniya M, Czaja AJ, , Parés A, Dalekos GN, Krawitt EL, et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 2008;48:169-176. [CrossRef]
- Hiejima E, Komatsu H, Sogo T, Inui A, Fujisawa T. Utility of simplified criteria for the diagnosis of autoimmune hepatitis in children. J Pediatr Gastroenterol Nutr 2011;52:470-473. [CrossRef]
- Manns MP, Czaja AJ, Gorham JD, Krawitt EL, Mieli-Vergani G, Vergani D, et al. Diagnosis and management of autoimmune hepatitis. Hepatology 2010;51:2193-2213. [CrossRef]
- Vergani D, Mieli-Vergani G. Pharmacological management of autoimmune hepatitis. Expert Opin Pharmacother 2011;12:607-613. [CrossRef]
- Kerkar N, Annunziato RA, Foley L, Schmeidler J, Rumbo C, Emre S, et al. Prospective analysis of nonadherence in autoimmune hepatitis: a common problem. J Pediatr Gastroenterol Nutr 2006;43:629-634 [CrossRef]
- Alvarez F, Ciocca M, Canero-Velasco C, Ramonet M, de Davila MT, Cuarterolo M, et al. Short-term cyclosporine induces a remission of autoimmune hepatitis in children. J Hepatol 1999;30:222-227 [CrossRef]
- Manns MP, Woynarowski M, Kreisel W, Lurie Y, Rust C, Zuckerman E, et al. Budesonide induces remission more effectively than prednisone in a controlled trial of patients with autoimmune hepatitis. Gastroenterology 2010;139:1198-1206 [CrossRef] [PubMed]
- Woynarowski M, Nemeth A, Baruch Y, Koletzko S, Melter M, Rodeck B, et al. Budesonide versus prednisone with azathioprine for the treatment of autoimmune hepatitis in children and adolescents. J Pediatr;163:1347-1353 e1
- Mieli-Vergani G, Vergani D. Budesonide for juvenile autoimmune hepatitis? Not yet. J Pediatr;163:1246-1248. [CrossRef] [PubMed]
- Aw MM, Dhawan A, Samyn M, Bargiota A, Mieli-Vergani G. Mycophenolate mofetil as rescue treatment for autoimmune liver disease in children: a 5-year follow-up. J Hepatol 2009;51:156-160. [CrossRef] [PubMed]
- Mitchell SA, Bansi DS, Hunt N, Von Bergmann K, Fleming KA, Chapman RW. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology 2001;121:900-907 [CrossRef] [PubMed]
- Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009;50:808-814 [CrossRef] [PubMed]
- Liberal R, Longhi MS, Grant CR, Mieli-Vergani G, Vergani D. Autoimmune hepatitis after liver transplantation. Clin Gastroenterol Hepatol 2012;10:346-353. [CrossRef] [PubMed]
- Scalori A HM, Hadzic N, Vergani D, Mieli-Vergani G. Outcome and survival in childhood onset autoimmune sclerosing cholangitis and autoimmune hepatitis: a 13 years follow-up study Hepatology 2007;46(Suppl 1):555A.
DOI: https://doi.org/10.7199/ped.oncall.2014.53
|
| Cite this article as: | | Liberal R, Vergani D, Mieli-Vergani G. AUTOIMMUNE LIVER DISEASE. Pediatr Oncall J. 2014;11: 61-64. doi: 10.7199/ped.oncall.2014.53 |
|