Sunil Jondhale1, Tushar Jagzape2, Anil Kumar Goel3, Kavita Tiwari4, Tripty Singh1, Manas Ranjan Sahoo1.
1Associate Professor, Pediatrics, All India Institute of Medical Sciences (AIIMS), Raipur, Chhattisgarh, India,
2Additional Professor, Pediatrics, All India Institute of Medical Sciences (AIIMS), Raipur, Chhattisgarh, India,
3Professor, Pediatrics, All India Institute of Medical Sciences (AIIMS), Raipur, Chhattisgarh, India,
4Senior Resident, Pediatrics, All India Institute of Medical Sciences (AIIMS), Raipur, Chhattisgarh, India.
ADDRESS FOR CORRESPONDENCE
Dr. Sunil Jondhale, D 423, 4th Floor, Hospital D block, Department of Pediatrics, AIIMS, Raipur and Chhattisgarh. India-492099.
Email: dr.sunjon358@yahoo.com | | Abstract | | One of the rare but a common symptom of some genetic diseases presenting in Pediatric age group is silvery hair. These disorders termed as “silvery hair syndrome” consist of Griscelli syndrome (GS), Chediak-Higashi syndrome (CHS) and Elejalde disease. Among these syndromes primary immunodeficiency is present only in CHS and GS type II. CHS which is characterized by severe defect in phagocytic function leading to recurrent infections with pyogenic organism, silver blond hair, partial oculocutaneous albinism, progressive motor or sensory neurologic defects and bleeding tendencies is a rare autosomal recessive defect. Griscelli syndrome 2 is also a rare disorder with autosomal recessive inheritance associated with dilution of pigments in skin, hair, splenohepatomegaly, immune and neurological dysfunction and pancytopenia. Because of overlapping clinical presentation, they closely mimic each other. Hence here we are focusing on differentiating these two disorders and highlighting the diagnostic dilemma. We report two cases which were referred to our tertiary care center of central India with varied clinical manifestations and complications, sharing a common feature of silvery hair. | | | | Keywords | | Silvery Hair, Immunodeficiency, Chediak-Higashi syndrome, Griscelli syndrome | | | | Introduction | | Silvery hair is a common feature of rare genetic immunodeficiency disorders which include Chediak‑Higashi syndrome (CHS), Griscelli Syndrome type II. They usually present in pediatric age group and inherited as an autosomal recessive disorder. Both disorders are characterized by partial albinism with silvery blonde hair, severe phagocytic immunodeficiency and may have invariable neurological dysfunction. Patients with these disorders have tendency to develop severe infections and recurrent life-threatening accelerated form of hemophagocytic lymphohistiocytosis. Here we are reporting two cases with silvery hair which were referred to our center with varied clinical presentations and complications. | | | | Case Report | Case 1
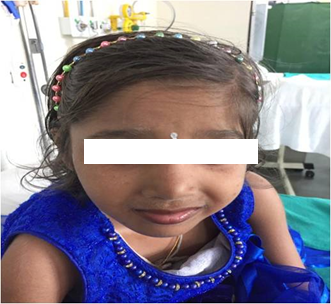
An 8-year-old developmentally normal female born of non-consanguineous marriage and had an uneventful antenatal and perinatal history presented with chief complain of fever, generalized weakness of all limbs and difficulty in breathing for eight days. She had a history of recurrent febrile illnesses in past requiring admissions. On admission, patient was febrile with moderate respiratory distress (RR40/min). She was severely underweight and stunted. On head-to-toe examination she was found to have grey hair on her head, eyebrows and eyelashes, her skin color appeared normal but she was fairer in comparison to other family members (Figure 1). On systemic examination she had bilateral decrease air entry with intercostals retractions with hepatosplenomegaly.CNS examination revealed weakness (proximal > distal) in all her limbs. Fundus examination was normal.
Provisional diagnosis of community acquired pneumonia with Guillain-Barre Syndrome (GBS)with suspected primary immunodeficiency was kept. She was started on oxygen and intravenous antibiotics. Bilateral diffuse heterogeneous opacities were seen in the Chest X-ray. CECT chest showed sub segmental atelactasis in basal segments of right and left lower lobe. In view of quadriparesis, EMG and NCV were done. EMG showed neurogenic changes in muscles. NCV showed axonal motor sensory radiculoneuropathy in both upper and lower limbs for which ivig was given suspecting GBS.USG abdomen showed hepatosplenomegaly with normal architecture. CBC showed pancytopenia (Hb of 10.2, TLC of 3500 with 64% neutrophils, 28% lymphocytes and Platelet count of 80000). Bone marrow examination showed normocellular marrow with no hemophagocytes. Tuberculosis workup was negative. Lab parameter like RFT, urine R/M and thyroid profile, serum ferritin, fibrinogen and triglyceride were in normal range. Liver function tests revealed mild elevation of enzymes with decreased albumin. Immunoglobulin assay showed elevated IgG (immunoglobulin levels total IgA 3.67, total IgG 14.20, total IgM 1.09). Flow cytometry showed elevated CD3 and CD4 with low CD8 and CD19 (absolute CD3 count-495, absolute CD4 count-335, absolute CD8 count-144, absolute CD19 count-32).On light microscopy of the hair, large clumps of melanin with irregular distribution were seen. Based on the history, clinical examination and laboratory parameters, differential diagnosis of Griscelli Syndrome type II(GS2) and Chediak-Higashi Syndrome(CHS)with GBS was made. Hair microscopy findings and failure to demonstrate inclusion in the granulocytes on peripheral blood examination ruled out CHS. Genome exon sequencing report was found positive for RAB27A mutation at intron 3 location suggestive of GS2(OMIM # 607624). Gradually, child improved in terms of respiratory distress as well as proximal muscle weakness. Hospital acquired infection was treated accordingly. Patient was referred to centre with BMT facility.
Figure 1. Griscelli syndrome type 2.
Case 2
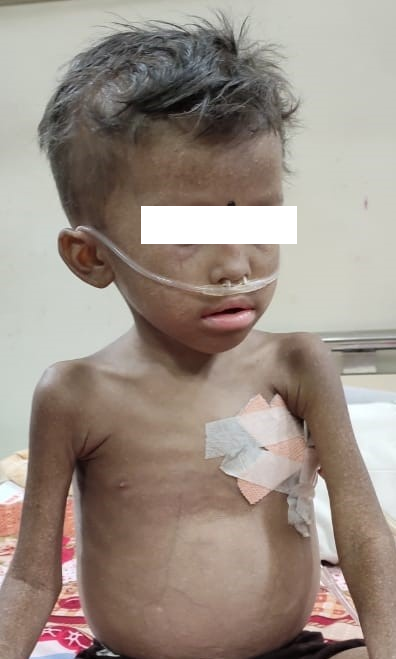
A 5 years old female child, 2nd born of a non -consanguineous marriage, developmentally normal, presented with intermittent high-grade fever for 1 year, with bilateral ear discharge for 5 months, cough and fast breathing for 1 month, epistaxis and gum bleeding for 1 day. She had past history of generalized skin lesions in the form of hypo-pigmented spots started appearing at the age of 1-month, recurrent episodes of chest infections, skin pustules and unprovoked generalized tonic-clonic seizure twice. There was a history of early neonatal death of elder female sibling with unknown cause. On examination patient was tachypneic, excessively fair skin in compare to other family members, moderately malnourished with weight/height between -2 to -3 SD with pallor, silvery grey sparse scalp hair, sparse eyebrows, nystagmus and pitting edema of both feet extending upto calf region. There were multiple hypo-pigmented macules evenly distributed throughout the body (Figure 2). Fundus examination revealed pigmentary changes. Abdomen was distended with firm, hepatomegaly (4 cm under right SCM) and firm splenomegaly (9 cm under left SCM)with no free fluid. On auscultation, there was decrease air entry in left hemithorax with increase dullness. No other abnormality was detected on rest physical examination.
Investigations revealed bicytopenia with Hb of 5.3 gm%, platelet of 15,000/micron mm with TLC of 20,000/micron mm with N30%, L62%. There were giant cytoplasmic granules in leucocytes. Markers for HLH were normal. CXR showed left massive pleural effusion and USG was suggestive of hepatosplenomegaly with organized left pleural collection. TFT, RFT, Tuberculosis workup, blood and urine culture were normal. Pleural fluid culture showed MSSA. Immunoglobulin assay was suggestive of decreased Immunoglobulin G levels. Sparse small sized clumps of melanin which were regularly spaced were seen when the hair shaft was examined under the microscope. Bone marrow examination was normal. Genetic analysis confirmed homozygous single base pair duplication in exon 5 of the Lystgene suggestive of Chediak Higashi Syndrome.
Patient was treated with intravenous antibiotics, right sided ICD drainage for empyema,supportive management and dietary rehabilitation. Patient was referred for bone marrow transplant.
Figure 2. Chediak Higashi Syndrome.
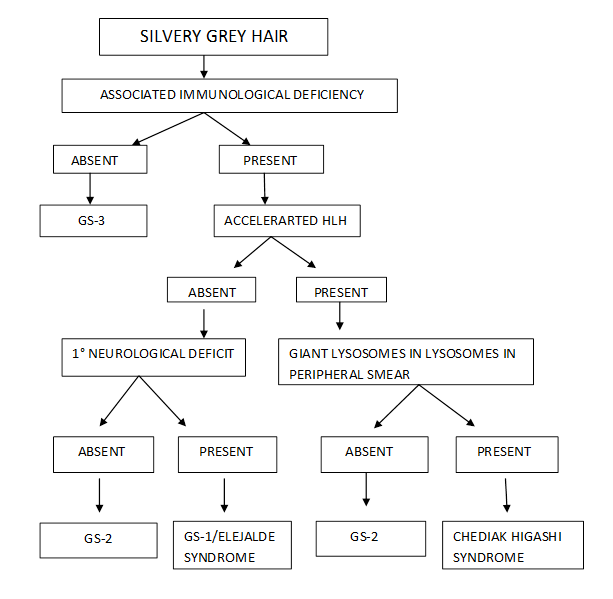
| | | | Discussion | The “silvery hair syndrome” constitute of three closely related autosomal recessive disorder of primary immunodeficiency syndromes which include Griscelli syndrome type II, Chediak Higashi Syndrome and Elejalde syndrome. These similar diseases can however be, differentiated from each other on basis of presence of immunodeficiency, neurological involvement, accelerated hemophagocytosis, peripheral smear finding and finally by genetic testing (Figure 3).
Figure 3. Approach to children with silvery Hair with Immunodeficiency12.
Griscelli syndrome (GS) is multisystem disorder with known three subtypes (GS1, GS2, GS3), according to location of the genetic mutation (Myosin VA, Ras related protein Rab-27A, melanophilinMLPH respectively).1 All the three types of GS have grayish hair in common. GS usually affects young children in the age group of 4 months to 4 years with no sex predilection.2 Griscelli syndrome type 1 (GS1) primarily presents with neurological involvement as development delay, seizures, intellectual disability and eye abnormality, without immune dysfunction. Elejalde syndrome closely mimics GS type 1.3 Griscelli syndrome type 2 (GS2) (also known as "partial albinism with immunodeficiency”)characterized by variable cutaneous albinism, silver colored metallic looking hair, repeated viral or bacterial infections, low neutrophil and platelet count with varying degrees of immunologic and secondary neurologic dysfunctions. Griscelli syndrome type 3 (GS3) does not need any treatment as it does not have any immunological or neurologic involvement and only has hypomelanosis.4
Out of the three types of GS, type 2 is the commonest. Till date approximately 103 cases have been reported across the world in Medical literature,5 which includes 13 cases from India.4,6 The primary immunodeficiency in GS2 patients is due to an impairment of T-cell cytotoxic activity and function of natural killer cells, which in turn leads to increased susceptibility to frequent infections. The neurological deficit in GS2 is secondary to the infiltration and proliferation of leukocytes in the brain unlike GS-1 which has primary CNS involvement.7
Chediak‑Higashi syndrome (CHS) is a rare autosomal recessive disorder characterized by severe defect in phagocytic function leading to recurrent pyogenic infections, partial oculocutaneous albinism (mild pigment dilution), silver blond hair, progressive motor or sensory neurologic defects and bleeding tendencies.8 Defective degranulation of the neutrophils increases the susceptibility of the patients to infections. The patients has progressive neutropenia as well as abnormalities in natural killer (NK) function. These patients have tendency to develop a life-threatening form of hemophagocytic lymphohistiocytosis. Failure of decussation of the optic and auditory nerves leads to neurologic deficits. Finding of large inclusions on microscopy in all nucleated blood cells is the diagnostic of CHS. Though frequent infections and neuropathy are common other symptoms and signs vary considerably. Skin, mucous membranes and respiratory tract are the common sites of infections. Affected children are susceptible to both gram positive and negative bacterias as well as fungi, the commonest among them being Staphylococcus aureus.8,9
In order to differentiate between CHS and GS 2 and to reach a correct diagnosis evaluation of the peripheral smear and microscopic examination of skin and hair shaft is required along with immunological workup (Table 1). Both the syndromes may prove fatal as due to immunological impairment the patients are at high risk of developing accelerated hemophagocytic syndrome. Fortunately, both our patients did not have hemophagocytosis till this visit. Diagnosis was made on the basis of clinical features, laboratory findings and genetic testing. Early recognition of patients with CHS andGS-2 and prompt intervention with bone marrow transplant, which is the only curative treatment for CHS and GS-2.10,11 The prognosis of both GS2 and CHS is poor, with mortality in early childhood due to HLH and its complications. Palliative management for both includes treatment of associated infections along with immunomodulatory therapy for accelerated phase (high dose methylprednisolone, intrathecal methotrexate, etoposide, cytosine arabino-side and prednisone or cyclosporine, atg and steroids).11,12 Treatment options are limited and the only definitive treatment is BMT. The prognosis is good if BMT is done before the setting in of accelerated phase of hemophagocytosis.
Table 1. Comparative differentiation of CHS and GS type 2.
| |
Chediak-Higashi syndrome |
Griscelli Syndrome type 2 |
| Neurological dysfunction |
Common and primary |
Uncommon and secondary |
| Bleeding tendency |
Common |
Uncommon |
| Ocular involvement |
Common |
Uncommon |
| Accelerated phase HLH |
Common, early |
Uncommon, late |
| Giant cytoplasmic granules on PS |
Present |
Absent |
| Light microscopy of hair |
Regularly arranged small clumps of melanin |
Irregularly arranged small and large clumps of melanin |
| Histopathology of skin |
Large melanosomes in both melanocytes and keratinocyte |
Excess pigmentation of melanocytes in basal layer with hypo-pigmentation of adjacent keratinocyte |
| | | | Conclusion | | These two clinical cases explain the need of awareness of this uncommon entity which if diagnosed late may prove fatal for the patient. Prompt initiation of supportive treatment followed by early bone marrow transplant is essential for the survival of such patients. Knowledge of differential diagnosis of silvery hair syndrome is alone sufficient to give the clue and to investigate such patients accordingly. Antenatal diagnosis and genetic counseling of the parents of such patients should be focused upon in subsequent pregnancies. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Griscelli C, Durandy A, Guy-Grand D et al . A syndrome associating partial albinism and immunodeficiency. Am J Med 1978;65:691-702. [CrossRef]
- Dotta L, Parolini S, Prandini A et al.Clinical, laboratory and molecular signs of immunodeficiency in patients with partial oculo-cutaneous albinism. Orphanet J Rare Dis. 2013; 8:168. [CrossRef] [PubMed] [PMC free article]
- Inamadar AC, Palit A. Silvery hair with bronze-tan in a child: A case of Elejalde disease. Indian J Dermatol VenereolLeprol2007;73:417-9. [CrossRef] [PubMed]
- Singh A, Garg A, Kapoor S et al. An Indian boy with griscelli syndrome type 2: Case report and review of literature. Indian J Dermatol 2014;59:394‑7. [CrossRef] [PubMed] [PMC free article]
- Meschede IP, Santos TO, Izidoro‑Toledo TC et al. Griscelli syndrome‑type 2 in twin siblings: Case report and update on RAB27A human mutations and gene structure. Braz J Med Biol Res 2008;41:839‑48. [CrossRef] [PubMed]
- Rajyalakshmi R, Chakrapani RN. Griscelli syndrome type 2: A rare and fatal syndrome in a South Indian boy. Indian J Pathol Microbiol. 2016; 59: 113-116.
- Weitzman S. Approach to hemophagocytic syndromes. Hematology Am Soc Hematol Educ Program 2011;2011:178‑83. [CrossRef] [PubMed]
- Shiflett SL, Kaplan J, Ward DM. Chediak-Higashi syndrome: a rare disorder of lysosomes and lysosome related organelles. Pigment Cell Res. 2002;15:251-57. (CHS). [CrossRef] [PubMed]
- L. Dotta, S. Parolini, A. Prandini, et al.Clinical, laboratory and molecular signs of immunodeficiency in patients with partial oculo-cutaneous albinism. Orphanet J Rare Dis 2013;8:168. [CrossRef] [PubMed] [PMC free article]
- Cesaro S, Locatelli F, Lanino E, et al. Hematopoietic stem cell transplantation for hemophagocytic lymphohistiocytosis: A retrospective analysis of data from the Italian Association of Pediatric Hematology Oncology (AIEOP). Haematologica. 2008; 93:1694-1701. [CrossRef] [PubMed]
- Gürgey A, Sayli T, Günay M, et al. High-dose methylprednisolone and VP-16 in treatment of Griscelli syndrome with central nervous system involvement. Am J Hematol 1994; 47:331-338. [CrossRef] [PubMed]
- Durrani S, Chicka M, Afroze B. Griscelli syndrome type 2 - a case report and clinical approach to silver blonde hair. Egypt J Med Hum Genet. 2016;17(2):229-232. [CrossRef]
DOI: https://doi.org/10.7199/ped.oncall.2023.43
|
| Cite this article as: | | Jondhale S, Jagzape T, Goel A K, Tiwari K, Singh T, Sahoo M R. Silvery Hair with Immunodeficiency: A Comparative clinical brief of Griscelli Syndrome Type II and Chediak Higashi Syndrome. Pediatr Oncall J. 2023;20: 149-153. doi: 10.7199/ped.oncall.2023.43 |
|