Abstract
Congenital adrenal hyperplasia comprises a group of autosomal recessive disorders characterized by enzyme blocks at various levels of adrenal steroid synthesis resulting in decreased production of adrenal hormones, mainly cortisol and aldosterone. the substrates which accumulate proximal to the defective enzyme lead to excess production of sex steroids. 21 hydroxylase deficiency constitutes about 95% of CAH and classic forms present in the neonatal period. They require life long supplementation with hydrocortisone and the majority (salt-wasting form) requires fludrocortisone also. The neonatal screening program helps in the early detection of these cases. Antenatal treatment with steroids is experimental at present.
Table 1: Pathways of steroid biosynthesis
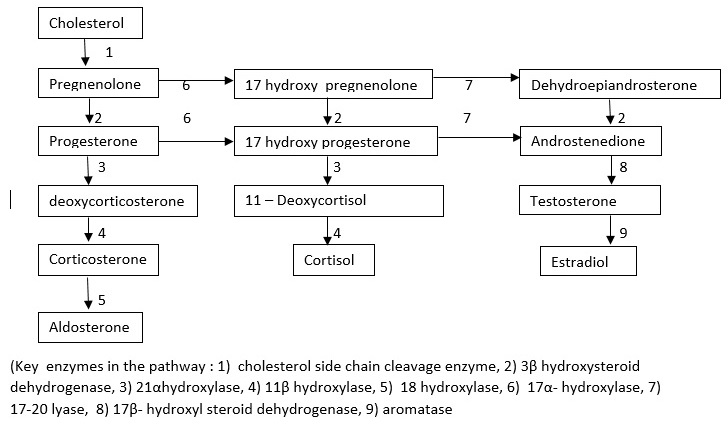
Introduction
Congenital Adrenal Hyperplasia is a group of autosomal recessive disorders that block the pathway of adrenal steroidogenesis at various steps. This condition creates adrenal insufficiency (defective production of glucocorticoids and mineralocorticoids). In response to adrenal insufficiency, the pituitary gland produces an increased amount of ACTH and POMC which stimulate the steroidogenesis pathway. This process results in hypertrophy and hyperplasia of adrenal glands, hence these conditions are collectively called congenital adrenal hyperplasia. The accumulated steroid precursors proximal to the block are converted to sex steroids. Clinical features of these groups of disorders arise from the deficiency of the steroid end products (mineralocorticoids and glucocorticoids) and also from the accumulating precursors proximal to the blocked step and their end products, formed due to the activation of alternate pathway. The main types of CAH are lipoid CAH, 3ß hydroxysteroid dehydrogenase deficiency (3? HSD), 17- a hydroxylase deficiency, 21 hydroxylase deficiency, and 11ß hydroxylase deficiency. 21 hydroxylase deficiency constitutes 95% of all cases of CAH, followed by 11-ß hydroxylase deficiency (4%). Other conditions are relatively rare.
21 Hydroxylase Deficiency
21 hydroxylase deficiency is due to mutations in the CYP21A2 gene and is one of the most common inborn errors of metabolism. Because of newborn screening programs, cases are being diagnosed in the newborn periods and due to efficient management, the majority of cases are reaching adulthood.
In the severest form, there is a complete block at the level of conversion of progesterone to deoxycorticosterone (DOC) which results in aldosterone deficiency causing severe hyponatremia, hyperkalemia, and acidosis. Block at the level of 17 hydroxyprogesterone to 11 deoxycortisol results in reduced cortisol production resulting in hypoglycemia. If this condition is not diagnosed in a neonate, associated hypotension, shock, and cardiovascular collapse result in the death of the affected child.
In utero, this inappropriately produced testosterone causes varying degrees of virilization of external genitalia in a girl child. The degree of virilization ranges from mild clitoromegaly with or without fusion of posterior labial folds to complete labioscrotal fusion with urethra situated in the enlarged clitoris. They present with atypical genitalia and create problems in sex assignment at birth. They have normally developed uterus, fallopian tubes, and ovaries.
Table 3: Serum 17 – hydroxyprogesterone levels in normal children (Adapted from Harriet lane)
| Age |
Baseline (ng/d L) | 60 minute post ACTH stimulation (ng/dL) |
| Cord blood | 2000 | |
| Neonate (>48 hours) | 420 | |
| Infancy (1-12 months) | 11-170 | 85-465 |
| 1-5 yr | 4-115 | 50-350 |
| 6-12 yr | 7-69 | 75-220 |
| Adolescents boys(Tanner ii,iii) | 12-130 | 69-310 |
| Adolescent boys (Tanner iv, v) | 51-190 | 105-230 |
| Adolescent girls (Tanner ii.iii) | 18-220 | 80-420 |
| Adolescent girls (Tanner iv,v) | 36-200 | 80-225 |
Congenital Adrenal Hyperplasia - Incidence
Neonatal screening for CAH with 17 OHP assay have shown that the incidence of classic CAH is 1 in 14,000. Even though exact data are not available, the incidence of non-Classic CAH is much higher.
Congenital Adrenal Hyperplasia - Clinical Presentations
In CAH, the clinical presentation is variable depending upon the severity of the limiting enzyme deficiency. Based on the clinical spectrum, this disease is classified into 3 groups. This classification is arbitrary because they just represent the different clinical presentation of the same disease
- Salt wasting CAH
- Simple virilizing CAH
- Non classic CAH
Salt Wasting CAH
This condition is caused by a complete deficiency of the enzyme 17 hydroxylase (P450 c 21) and results in a complete block in the synthesis of glucocorticoids and mineralocorticoids. Girls present with virilization at birth and this is the most common form of 46 XX DSD. These babies often require resuscitation for the cardiovascular collapse, hypoglycemia, acidosis, and electrolyte abnormalities at presentation. Mineralocorticoids and glucocorticoids are replaced. They need periodic monitoring and titration of glucocorticoids and mineralocorticoids. Girls need surgical correction of their atypical genitalia. Many a time boys are diagnosed late during their salt-losing crisis (5-15 day of life) or not diagnosed at all resulting in neonatal death due to the wrong diagnoses like sepsis or hypoglycemia.
Simple Virilizing CAH
A simple virilizing form of CAH is a milder variety when compared to the salt-losing variety. In girls, the presence of atypical genitalia leads to early detection. But males (unless detected by newborn screening) may present later when they develop premature pubic, axillary, or facial hair or notice increase in phallic growth, inappropriate for their age. Their testicular volume is pre-pubertal, differentiating from central precocious puberty, in which testicular volume increases (>3 ml) due to gonadotropin stimulation. Their linear growth velocity will be above their peers, but they end up as short adults because their bone maturation advances faster resulting in an early fusion of epiphyses. In untreated cases, boys will have small testes, delayed puberty, and azoospermia because the adrenal androgens produce negative feedback to the hypothalamus, suppressing the pituitary gonadotropins. Once treatment is begun in childhood, this suppression is removed and this occasionally results in central precocious puberty. A high concentration of ACTH in a poorly managed child leads to the development of adrenal rests in the testes.
Non Classic CAH
This is a mild form of 21- hydroxylase deficiency. This condition is also called late-onset CAH. They may present in older children with hirsutism, virilization, acne, menstrual irregularities, or decreased fertility. They may just present as a normal child with elevated 17- OHP level to ACTH stimulation test (cryptic CAH) and high PRA (Plasma Renin Activity)
Table 2. ACTH stimulation test
- Basal value of 17 OHP and cortisol are measured
- Synthetic ACTH is administered IV (Dose 0.1 mg in NB, 0.15 mg up to 2 yrs, 0.25 mg in >2 years)
- Repeat values are measured at 60 minutes
- Results: in classical CAH, both basal (> 2000 ng/dL) and stimulated (5,000-10,000 ng/dL) will be elevated. In non- classical type, normal or mild elevation of basal 17-OHP with high stimulated levels.
- A complete adrenocortical profile helps to differentiate 21 hydoxylase deficiency from other type of CAH
Other Types of CAH
Compared to 21 hydroxylase deficiency, other varieties of CAH are rare.
CAH due to 11ß hydroxylase may also present with a salt-wasting crisis in the newborn period since the amount of deoxycorticosterone (DOC) produced is insufficient to meet the mineralocorticoid requirement in the newborn period. But the salt-wasting episode is less severe when compared with 21 hydroxylase deficiency. But mineralocorticoid requirement is less as the age advances and they develop hypertension.
Table 4 : Comparison of the potency of various steroid preparations
| Steroid |
Anti inflammatory effect |
Growth retarding effect |
Salt -retaining (mineralocorticoid) effect |
| Cortisol (hydrocortisone) |
1 |
1 |
1 |
| Cortisone |
0.8 |
0.8 |
0.8 |
| Prednisone/ prednisolone |
4 |
5 |
0.25 |
| Methyl prednisolone |
5 |
7.5 |
0.4 |
| Betamethasone |
25 |
|
0 |
| Dexamethasone |
30 |
80 |
0 |
| Fludrocortisone |
15 |
|
200 |
| Triamcinolone |
5 |
|
0 |
Congenital Adrenal Hyperplasia - Diagnosis
Estimation of 17- hydroxyl progesterone
Elevated 17 OHP level is the gold standard test of 21- hydroxylase deficiency. Both basal and stimulated levels are markedly elevated in salt-losing ad simple virilizing forms. Basal levels are usually >2000 ng/dL and they increase to >5000-10,000 ng/dL after ACTH. In milder late-onset variety, basal levels may be mildly elevated but the stimulated values will be high (1500-10,000 ng/dL). The ideal time of measurement of 17- OHP is before 8.00 AM
17 OHP level in a neonate
17 OHP levels decease in utero as the gestational age advances. Hence its levels are high in preterm. Levels are also higher in SGA babies and babies under stress (birth asphyxia, cardiac or pulmonary disease) resulting in false-positive results. Levels are high in cord blood (1000-3000 ng/dL). After birth, 17-OHP levels fall dramatically in the first 48 hours and reach a baseline value. Hence a falsely high value may be obtained if the measurement is done before 48 hours.
Congenital Adrenal Hyperplasia - Other Investigations
Usually, the cortisol response will be subnormal to ACTH stimulation. Basal plasma ACTH levels will be very high in classic forms of CAH but may be normal or have mild elevation in milder forms. Urinary excretion of 17- ketosteroids will be elevated, but this test is rarely used nowadays. While doing this test 24- hour collection is a must and concomitant measurement of creatinine excretion should be done.
Plasma Renin Activity
Plasma renin activity is an immunoassay of the amount of angiotensin 1 generated per ml of serum per hour at 37°C. Usually, PRA is detected twice, once in the morning after overnight supine position and again after the maintenance of upright posture for 4 hours. Levels of PRA will be high in children with CAH. Elevated PRA and a reduced ratio of aldosterone to PRA indicates impaired aldosterone synthesis and can differentiate salt-wasting variety from simple virilizing forms of CAH.
Newborn Screening
CAH is ideal for newborn screening because this condition is relatively common and unless picked up in the newborn period and effectively managed, many succumb without a proper diagnosis. Girs may be picked up because of their atypical genitalia.
Measurement of 17-hydroxyprogesterone (17-OHP) level is the screening tool used in newborn screening programs. 17- OHP levels are normally high at birth. Its level falls during the first few days after birth in normal babies but progressively increases in children with CAH. Hence accuracy is poor in the first few days. Preterm, sick babies, SGA, and stressed neonates have higher 17-OHP levels and may give false-positive results. Hence the screening test is usually done after 48 hours. Once a positive result is obtained, it should be confirmed by a second assay.
Molecular Genetic Screening
In places where facilities are available, CYP21A2 mutations can be detected in DNA extracted from the dried blood spots in the neonatal period itself.
Glucocorticoid Supplementation
The daily requirement of cortisol for effective suppression of ACTH and adrenal androgen production is about 10-15 mg/m2. Neonates and newly diagnosed patients require a higher dose (15-20 mg/m2) initially to suppress their hyperactive CRH-ACTH- adrenal axis. Various glucocorticoid dose equivalents are based on their anti-inflammatory properties, and the growth suppressing effect does not match with anti-inflammatory properties. Dexamethasone and prednisolone have powerful growth suppressant effects compared to hydrocortisone. Hence these drugs are better avoided in children. But it can be used in adults where epiphyseal fusion has already completed.
Mineralocorticoid Therapy
Its use is well established in a salt-losing variety of CAH. In simple virilizing types also it is advised because in these children the dose of glucocorticoids can be reduced and hence the growth suppression and weight gain can be avoided. The only mineralocorticoid available at present is fludrocortisone. Neonates require larger doses of fludrocortisone (0.15-0.3 mg/day) compared to older children (0.05- 0.15 mg/day).
For the action of mineralocorticoids, there should be an adequate amount of sodium in the renal tubule. Hence additional salt supplementation (1-2 g of NaCl/day) is required in early infancy.
Table 5 : Maintenance therapy in children with CAH
| Drug |
Total daily dose |
Daily distribution |
| Hydrocortisone |
10- 15 mg/m2 |
3 times |
| Fludrocortisone |
0.05-0.2 mg |
1-2 times |
| Sodium chloride supplementation |
1-2 grams (17-34 mEq/day) in infancy |
Divided in several feedings |
Congenital Adrenal Hyperplasia - Follow Up
Effective management requires close monitoring. Growth measurements should be made at 4-6 monthly intervals. Bone age should be assessed every year. Blood pressure is recorded in each visit. Measurement of serum electrolytes (Na, k) should be performed in each visit. PRA and 17 OHP measurements are done at least once a year. Attempts to completely normalize 17- OHP levels result in overtreatment. Children with good treatment protocols have mildly elevated 17-OHP levels. Dose adjustment is made based on overall clinical profile and anthropometric measurements rather than based on the 17-OHP value alone.
Congenital Adrenal Hyperplasia - Treatment During Stress
Children with CAH are unable to produce enough cortisol in response to stressful situations like febrile episodes, diarrhea, surgery, or trauma. They require an increased dose of glucocorticoids (hydrocortisone) in such situations. When a pharmacological dose of hydrocortisone is given, mineralocorticoids are not required because the pharmacological dose of hydrocortisone has enough mineralocorticoid action. Patients should be given glucose and electrolyte supplementation and should never be allowed to fast. The initial stress dose of hydrocortisone is 60- 100 mg/m2. Subsequent doses are given as 3-4 times the maintenance dose of hydrocortisone per day, divided every 6 hours.
Table 6: Initial stress dose of hydrocortisone in different age groups
| Age groups |
Weight groups (kg) |
Dose of hydrocortisone (mg) |
| < 6 months |
< 7 |
25 |
| 6 months-2 years |
8-12 |
50 |
| 3-10 years |
13-30 |
75-100 |
| > 10 years |
>30 |
100-200 |
Congenital Adrenal Hyperplasia - Surgical Options
Surgical reconstruction should be considered in infancy itself in a severely virilized child (Prader stage >3). In less virilized children clitoroplasty and vaginoplasty can be done at a later age.
Congenital Adrenal Hyperplasia - Prenatal Treatment
Prenatal treatment continues to remain as experimental. This method is based on the fact that suppression of fetal adrenal androgen in CAH can be achieved by administering glucocorticoids to the mother. This will reduce the genital virilization of the affected girl child and reduce the need for genital reconstructive surgery. The drug used is dexamethasone since this drug is not inactivated by placental 11ß- HSD2. The treatment must be started at 6-7 weeks of gestation. Fetal DNA is obtained by chorionic villus sampling at 10-12 weeks. The therapy is continued only if the fetus is a girl.
This treatment results in various maternal problems. These include weight gain, pedal edema, hypertension, striae, gestational diabetes, and mood swings. In a fetus, it may develop a cleft palate and other craniofacial abnormalities.
Table 7: presentation of different types of CAH
| Type |
Presentation |
Laboratory findings |
|
| Lipoid CAH |
Salt wasting crisis
Under virilized male
|
Low levels of all steroid hormones
Decreased response to ACTH
High ACTH & PRA |
Gluco and mineralo corticoid replacement
Salt supplementation
Estrogen replacement at puberty
Gonadectomy for male
|
| 3ß HSD |
Salt wasting crisis
Under virilized male |
High d 5 steroids
High ACTH & PRA |
Gluco and mineralo corticoid replacement
Salt supplementation
Genital surgical correction
|
| 21- Hydroxylase deficiency |
Salt wasting crisis
Virilized female |
High 17-OHP
High serum androgens
High ACTH & PRA |
Gluco and mineralo corticoid replacement
Salt supplementation
Surgical genital correction in girls
|
| 11 ß hydroxylase deficiency |
Virilized female
Hypertension |
High 11- Deoxy cortisol and Deoxy corticosterone
High 17- OHP
High ACTH, Low PRA
Hypokalemia
|
Glucocorticoid replacement
Surgical genital correction in girls
|
| 17 a hydroxylase deficiency |
Under virilized male
Hypertension |
High DOC, corticosterone,
High ACTH, low PRA
Hypokalemia |
Glucocorticoid administration
Surgical correction of genitalia and sex steroid replacement in boys
|
Treatment
The treatment of CAH is a double-edged sword. Overtreatment results in linear growth failure, hypertension, and cushingoid features. Undertreatment results in lack of effective suppression of adrenal androgen production resulting in early epiphyseal fusion and short stature.
Complications
Prolonged steroid therapy reduces bone mineral density. The problem is not high if the physiological replacement dose is administered. Age-appropriate Vitamin D and Calcium intake should be ensured in all children on treatment. Some children are prone to adrenal rest tumors. They are bilateral and are >2 cm in diameter, hence not palpable. They are benign and related to suboptimal therapy. They decrease in size after receiving optimal treatment. Irregular menstrual cycles are common in adolescents.
1. Saroj Nimkarn Maria I. New; Prenatal Diagnosis and Treatment of Congenital Adrenal Hyperplasia; Horm Res 2007;67:53–60.
2. Mimi S. Kim, Anna Ryabets-Lienhard, Mitchell E. Geffner; Management of Congenital Adrenal Hyperplasia in Childhood; Curr Opin Endocrinol Diabetes Obes. 2012 December ; 19(6): 483–488.
3. Christine M. Trapp, Phyllis W. Speiser, Sharon E. Oberfield; Congenital adrenal hyperplasia: an update in children, Curr Opin Endocrinol Diabetes Obes. 2011 June ; 18(3): 166–170.
4. Christine M. Trapp, Sharon E. Oberfield; Recommendations for Treatment of Nonclassic Congenital Adrenal Hyperplasia (NCCAH): an Update; Steroids. 2012 March 10; 77(4): 342–346.
5. Deborah P Merke, Prof. Dix P Poppas; Management of adolescents with congenital adrenal hyperplasia; Lancet Diabetes Endocrinol. 2013 December ; 1(4): 341–352.
6. Hershel Raff, Susmeeta T. Sharma, Lynnette K. Nieman; Physiological Basis for the Etiology, Diagnosis, and Treatment of Adrenal Disorders: Cushing’s Syndrome, Adrenal Insufficiency, and Congenital Adrenal Hyperplasia; Compr Physiol. 2014 April ; 4(2): 739–769.
7. Phyllis W. Speiser, Ricardo Azziz, Laurence S. Baskin, Lucia Ghizzoni; Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline.
8. Perrin C White; Congenital adrenal hyperplasia and related disorders. In Robert M Kliegman (ed) Nelson Textbook of Pediatrics; 20th edn (first South Asia Edition). Elsivier India Publication, New Delhi; 2016 2714-2722.
9. Anurag Bsajpai, PSN Menon; Copngenital adrenal hyperplasia. In Piyush Gupta (ed), PG textbook of Pediatrics, 1st ed, Jaypee Publishers, New Delhi; 2015; 2341- 2344.
10. Palany Raghupathy, Disorders of Adrenocortical Biosynthesis. In Meena P Desai (ed)Pediatric Endocrine Disorders, 3rd ed. Universities press. New Delhi; 2015:227-238.