Definition, Classification
Definition:
As in adults, pulmonary hypertension (PH) is present when the mean pulmonary artery pressure (mPAP) exceeds 25 mm Hg in term babies at sea level after 3 months of age.
In the context of congenital heart disease, it is important to emphasize that pulmonary hypertensive vascular disease (PHVD) may be present even when the mPAP is less than 25 mm Hg (e.g., in the low-flow situation of a cavopulmonary shunt when there is no subpulmonary ventricle). On the other hand, PHVD may not be present even when the mPAP is greater than 25 mm Hg (e.g., when there is a pulsatile source of pulmonary blood flow from a systemic-pulmonary artery shunt). Even though indexed pulmonary vascular resistance (PVRi) is not used in the definition of PH, it plays an important role in the diagnosis and management of PHVD in patients with congenital heart disease.
Pulmonary artery hypertension (PAH) is present when the mPAP exceeds 25 mm Hg, but the pulmonary artery wedge pressure is less than 15 mm Hg, though the PVRi exceeds 2 Woods Units/m2. When no underlying etiology for PAH is found, the condition is referred to as idiopathic pulmonary hypertension (IPAH).
Classification:
The WHO classification of adult PH was modified at the Fifth World Symposium for Pulmonary Hypertension, held in Nice, France. The revised clinical classification of PH groups patients into classes based on purported etiologic mechanisms (Table 1). Group 1 includes patients with pulmonary arterial hypertension secondary to genetic mutations, drugs, toxins, and infections. It also includes patients with idiopathic and heritable PAH. Separate subgroups include those with veno-occlusive disease and persistent pulmonary hypertension of the newborn. Group 2 includes patients with PH secondary to left heart disease. Patients with PH due to lung diseases, with or without hypoxia comprise Group 3. Group 4 includes patients with chronic thromboembolism. PH associated with unclear but multifactorial mechanisms forms Group 5.
Specifically, with regard to congenital heart disease, the Nice modification recognizes 4 types associated with PAH (Table 2). Type 1 includes patients with classic Eisenmenger syndrome and right-to-left shunting. Type 2 includes patients with PHVD secondary to a large left-to-right shunt. Type 3 includes patients with PAH and co-incidental CHD, while type 4 includes patients with repaired CHD who develop PAH in the post-operative period. It is important to remember that patients without a subpulmonary ventricle, who do not meet the criteria for PH, remain at risk for pulmonary vascular disease, and are not represented in this classification.
The Nice classification is adult-focused, and may not truly reflect the heterogeneity of neonatal and pediatric PH. The pediatric task force of the Pulmonary Vascular Research Institute proposed the Panama Classification of PHVD in the pediatric population (Table 3), that highlights the phenotypic heterogeneity in this patient population.
Table 1: WHO Classification of Pulmonary Hypertension
- 1. PAH
- 1.1 Idiopathic
- 1.2 Heritable
- 1.3 Drug and toxin induced
- 1.4 APAH
- 1' PVOD and/or PCH
- 1.1" PPHN
- 2. PH due to left-sided heart disease
- 2.1 LV systolic dysfunction
- 2.2 LV diastolic dysfunction
- 2.3 Valvular disease
- 2.4 Congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathy
- 3. PH caused by lung disease or hypoxemia
- 3.1 Chronic obstructive pulmonary edema
- 3.2 Interstitial lung disease<
- 3.3 Other pulmonary diseases with mixed restrictive and obstructive pattern
- 3.4 Sleep-disordered breathing
- 3.5 Alveolar hypoventilation syndromes
- 3.6 Long-term exposure to high altitudes
- 3.7 Developmental lung diseases
- 4. Chronic thromboembolic disease
- 5. PH with unclear or multifactorial mechanisms
- 5.1 Hematological disorders: chronic hemolytic anemia, myeloproliferative disorders, splenectomy
- 5.2 Systemic disorders: sarcoidosis, pulmonary histiocytosis, lymphangioleiomyomatosis
- 5.3 Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders
- 5.4 Others: tumor obstruction, fibrosing mediastinitis, chronic renal failure, segmental PH
Table 2: Classification of PAH associated with congenital heart disease
- Eisenmenger syndrome
- Left-to-right shunts
- PAH with co-incidental CHD
- Post-operative PAH
Table 3: Panama Classification of PHVD in the Pediatric Population
- Prenatal or developmental PH vascular disease
- Perinatal pulmonary vascular maladaptation
- Pediatric cardiovascular disease
- Bronchopulmonary dusplasia
- Isolated pediatric pulmonary hypertensive vascular disease
- Multifactorial pulmonary hypertensive vascular disease in congenital malformation syndromes
- Pediatric lung disease
- Pediatric thromboembolic disease
- Pediatric hypobaric hypoxic exposure
- Pediatric pulmonary vascular diseases associated with other system disorders
Symptoms, Physical Examination
Symptoms:
PH may manifest with non-specific symptoms. In neonates and infants, frequently, the only symptom might be cyanosis perceived by the parents. In older children and adolescents, non-specific symptoms like effort intolerance, poor appetite and poor growth, nausea, and vomiting may be present. In some instances, the presentation may be more ominous in the form of exertional dyspnea or syncope. In most cases, a high index of suspicion prompts diagnostic testing. Patients with pulmonary hypertension are at risk for hypertensive crises, which are exacerbated by agitation and acidosis, and may result in chest pain, syncope, or even sudden death.
Physical Examination:
In neonates and infants with a patent foramen ovale and right-to-left shunting, systemic desaturation is present. This might also be present in older children with atrial communication. Rarely, clubbing of the nail beds is noted. While hepatomegaly and ankle edema is present in the older population, this is not likely to be seen in infants.
The cardiac examination is remarkable for a right ventricular heave and precordial bulge on inspection, a palpable second heart sound on palpation, and a loud second heart sound which is widely split (though not fixed) on auscultation.
Work-up of Pulmonary Hypertension
Work-up of pulmonary hypertension:
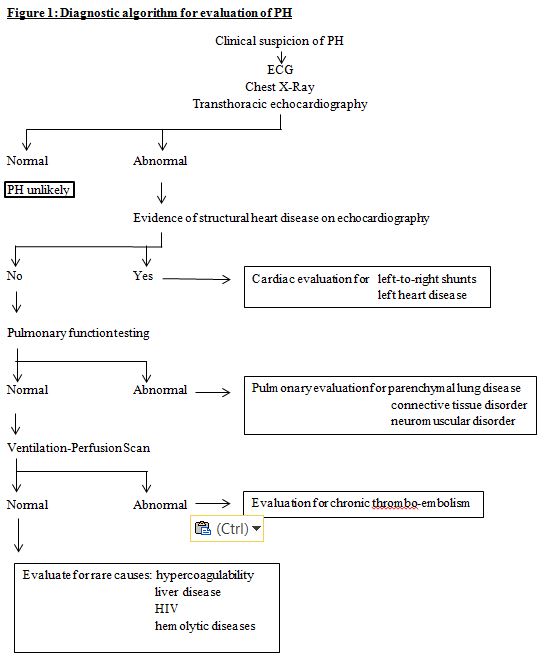
When a patient’s history, symptoms, and signs suggest a high likelihood of pulmonary hypertension, the work-up consists of a complete history (including family history and that of conditions likely to result in pulmonary hypertension) and investigations that not only confirm the diagnosis, but also help in identifying an etiologic factor, in quantifying the severity of the disease, and in judging the likely success of medical management. Ideally, this work-up is carried out in centers dedicated to the management of PH that have the necessary multidisciplinary expertise. Figure 1 depicts a diagnostic algorithm for the diagnosis of pediatric PH.
General laboratory studies: These include a complete blood count, serum chemistries, urinalysis, and BNP. A chest X-Ray in older children and adolescents might show pulmonary oligemia with the pruning of blood vessels. The cardiac apex is upturned, and the pulmonary notch is very prominent.
Cardiac investigations: These investigations help confirm the diagnosis, classify the severity, and assess the response to medical therapy. Non-invasive methods are useful in a longitudinal follow-up of pulmonary hypertension.
Figure1 - Diagnostic algorithm for evaluation of PH
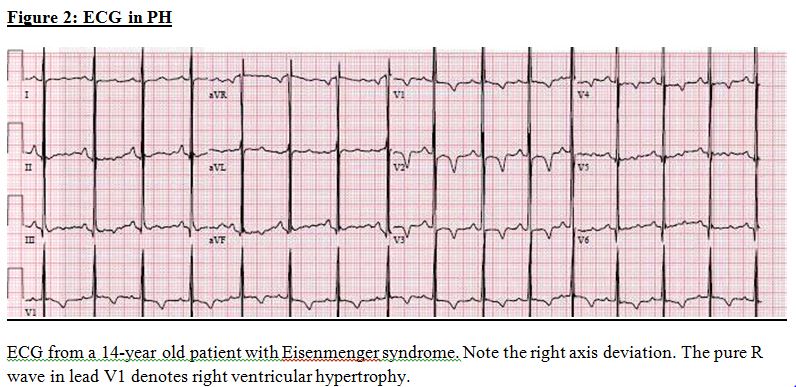
ECG: In established cases, right axis deviation, right ventricular hypertrophy, and right atrial enlargement are seen (Figure 2).
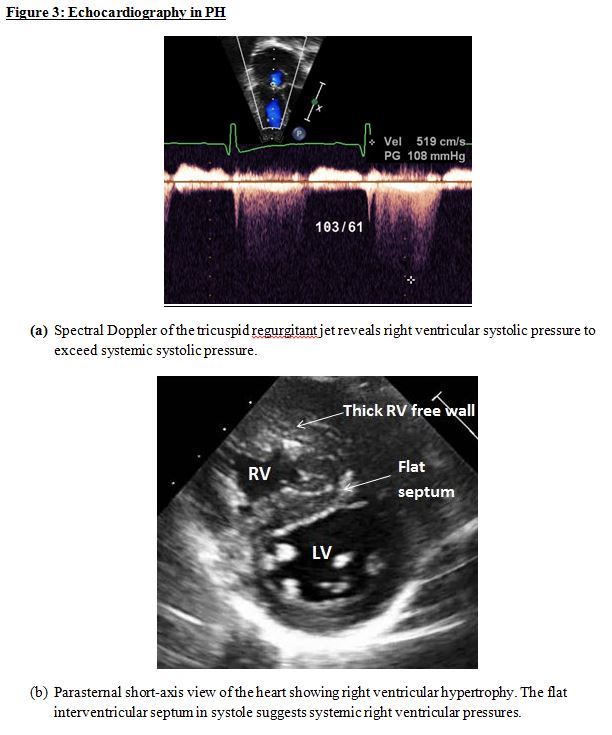
Echocardiogram: A transthoracic echocardiogram can rule out a congenital heart lesion that can result in PH when unrepaired. It can also rule out left-heart defects (pulmonary vein stenosis, mitral inflow defects, left ventricular outflow defects, and coarctation of the aorta) that can cause PH. When the spectral Doppler profile of the tricuspid or pulmonary regurgitant jet is of good quality, right ventricular systolic pressure can be determined non-invasively (Figure 3a). This is not only helpful for confirming the diagnosis of PH, but also in a longitudinal follow-up. Right ventricular hypertrophy (Figure 3b) and dilation, as well as systolic and diastolic function, can be assessed non-invasively. Three-dimensional echocardiography is a useful tool to assess right ventricular volumes. The left ventricular systolic and diastolic function can also be assessed.
ECG in PH
Cardiac Magnetic Resonance Imaging: CMR is the gold standard with respect to the assessment of right ventricular volumes and mass. While not as readily available or inexpensive as an echocardiogram, it might be valuable in the longitudinal follow-up of the right ventricle. In the setting of congenital heart disease, it is also useful in defining anatomy, especially in the setting of repaired heart disease wherein echocardiographic imaging windows might be suboptimal. It is useful in assessing differential flow to lung segments, as well as in assessing pulmonary thrombi.
Computed tomography: A CT Scan of the chest can be obtained in a short duration of time, and might obviate the need for anesthesia for imaging. Its value is primarily in the diagnosis of pulmonary thromboembolism, as well as in the differentiation of pulmonary venous-occlusive disease from pulmonary capillary hemangiomatosis.
Echocardiography in PH
Cardiac Catheterization: Catheterization is the gold standard of diagnosis of PH. The mPAP as well as the PVR can be assessed invasively. In infants and young children, where one is reasonably certain about the diagnosis of PH on the basis of non-invasive imaging, cardiac catheterization may not be done to avoid the morbidity of the procedure. In the setting of congenital heart disease, catheterization is valuable in assessing hemodynamics. The main utility of catheterization lies in our ability to carry out acute vasoreactivity testing (AVT) in the laboratory. The vasodilator agents used are oxygen, inhaled nitric oxide, and inhaled or intravenous prostacyclin analogs. This predicts the prognosis with the use of pulmonary vasodilators in the outpatient setting. The response to AVT also helps in determining operability in the setting of unrepaired congenital shunt lesions. Cardiac catheterization in patients with PH carries a significant risk of morbidity (acute hypertensive crises) and mortality, and should only be performed in centers that have experience with the management of PH.
Endurance testing: Both the 6-minute walk test and bicycle/treadmill ergometry are useful in the longitudinal follow-up of patients with PH.
Genetic testing: In the setting of a positive family history of PAH, genetic testing may be offered. BMPR2 has been identified as the primary pulmonary hypertension gene in the setting of HPAH. Genetic counseling in cases with a positive genotype is challenging given the incomplete penetrance of this gene and the possibility that a “second hit” explains the association of a positive genotype with the development of PH.
Management Of PH
Management of PH: PH should preferably be managed by specialized centers that have the multidisciplinary team and logistics to ensure appropriate testing and ongoing surveillance.
Pharmacotherapy:
Diuretics
Digoxin
Calcium channel blockers: Amlodipine, Nifedipine, Diltiazem. These agents are preferably used in children older than 1 year of age, as they are poorly tolerated in infancy.
Endothelin receptor antagonists: Bosentan, Ambrisentan.
Phosphodiesterase-5 inhibitors: Sildenafil, Tadalafil.
Prostacyclin analogs: Iloprost (inhaled), Treprostinil (subcutaneous), Epoprostenol (intravenous), Beraprost (oral).
Interventional therapy:
ASD creation, Palliative Potts shunt: These interventions aim to increase cardiac output at the expense of cyanosis.
Lung transplantation
Supportive measures:
Oxygen
Anticoagulation
Approach to treating a patient with PH:
The 2013 World Symposium on Pulmonary Hypertension arrived at a consensus regarding the algorithm towards the management of idiopathic or familial pulmonary artery hypertension. After the diagnosis of PH is confirmed, the patient is stabilized with diuretics, digoxin, anticoagulation, and oxygen.
Acute vasoreactivity testing (AVT) is the next step in deciding on optimal medical therapy. Patients who show a positive response to AVT should be treated with calcium channel blockers. If the response is sustained, calcium channel blockers should be continued. If there is no response to calcium channel blockers, therapy should be escalated to endothelin receptor antagonists or phosphodiesterase inhibitors.
Patients who show a negative response to AVT should be stratified into low and high-risk categories. Clinical evidence of right ventricular failure, the progression of symptoms on therapy, syncope, growth failure, WHO functional class III or IV, significant elevation of BNP, echocardiographic evidence of right ventricular enlargement, dysfunction or pericardial effusion are high-risk factors. On cardiac catheterization, a systemic cardiac index less than 2.5 L/min/m2, mPAP to mSAP ratio >0.75, RA pressure >10 mm Hg, and PVRi >20 Woods units are high-risk factors. Patients who do not have high-risk criteria should be initially treated with endothelin receptor antagonists or phosphodiesterase inhibitors. Those with high-risk markers should be initially treated with prostacyclin analogs, with a low threshold for considering combination therapy with endothelin receptor antagonists or phosphodiesterase inhibitors.
All patients should be reassessed at 3-month intervals to assess the efficacy of therapy and sustained improvement. In cases of clinical deterioration, combination therapy should be considered. In patients that are unresponsive to medical therapy, palliative procedures such as atrial septostomy or a Potts shunt should be considered. These patients should be evaluated for candidacy for a lung transplant.
Special clinical scenarios involving PH:
Persistent pulmonary hypertension of the newborn:
Use of inhaled nitric oxide, sildenafil, and prostacyclin analogs.
Supportive care with ventilation, and intravenous milrinone.
PH in the setting of structural heart disease:
Cardiac catheterization to measure PVRi.
If PVRi <6 Woods units, consider surgical repair.
If PVRi >6 Woods units, and positive response to AVT - consider surgical repair.
If PVRi >6 Woods units, and negative response to AVT - surgical repair is not beneficial. Medical treatment of PH, with a reassessment in 4-6 months.
1. Rabinovitch M. Pathophysiology of pulmonary hypertension. In: Moss and Adams’ Heart Disease in Infants, Children, and Adolescents: Including the Fetus and Young Adult – 8th Edition. Allen HD, Driscoll DJ, Shaddy RE, Feltes TF, Eds. Lippincott, Williams & Wilkins, Philadelphia, PA 2013. pp 1401-1432.
2. Ivy DD. Clinical management of pediatric pulmonary artery hypertension. In: Moss and Adams’ Heart Disease in Infants, Children, and Adolescents: Including the Fetus and Young Adult – 8th Edition. Allen HD, Driscoll DJ, Shaddy RE, Feltes TF, Eds. Lippincott, Williams & Wilkins, Philadelphia, PA 2013. pp 1433-1462.
3. Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009;54:S43-S54.
4. Cerro MJ, Abman S, Diaz G, et al. Functional classification of pulmonary hypertension in children: report from the PVRI Pediatric Taskforce, Panama 2011. Pulm Circ 2011;1:280-285.
5. Abman SH, Hansmann G, Archer SL, et al. Pediatric pulmonary hypertension: Guidelines from the American Heart Association and American Thoracic Society. Circulation 2015;132:2037-2099.
6. Ivy DD, Abman SH, Barst RJ, et al. Pediatric pulmonary hypertension. J Am Coll Cardiol 2013;62:D117-D126.