Introduction
Retinoblastoma is the most common primary tumor of the eye that arises from the nuclear layer of the retina and accounts for about 3% of the cancers occurring in children younger than 15 years of age.
Retinoblastoma can occur in both hereditary (40%) and sporadic (60%) forms. The hereditary disease includes those patients with positive family history and those patients who have inherited a germline mutation from an unaffected parent. It has been found associated with deletions or mutations of the "Retinoblastoma" gene on the q14band of chromosome 13. The germline form can manifest both as a unilateral or bilateral disease. However, most unilateral diseases are sporadic whereas all bilateral diseases are hereditary. But, unilateral disease in an infant is more likely to be a hereditary condition. 95% of the diagnosed cases of Retinoblastoma occur before the age of 5 years.
Pathophysiology And Genetics
The genetic basis of retinoblastoma has been well established. Loss of tumor suppressor function of the RB1 gene located on chromosome 13q14, by mutation or deletion leads to RB formation. Apart from this many other mutations may cause uncontrolled growth of immature retinal cells (retinoblastoma retinal progenitor cells) giving rise to this tumor. Recently, lower dietary intake of fresh fruits and vegetables in pregnant mothers has been linked with an increased incidence of unilateral, sporadic cases.
For a normal cell, to convert into an abnormal cell of retinoblastoma, two steps are involved. This is called "Knudson's two-hit hypothesis ". The First hit causes one allele to become abnormal. This alone will not cause retinoblastoma. The second "hit" would "knock off" the only remaining normal allele and hence would give rise to the tumor.
As with most other genetic conditions; based on when and where the mutation occurs, the presentation of the disease differs.
Sporadic non-heritable
(60%): If the genetic mutation is sporadic, wherein both "hits " affect a single somatic cell, it gets mutated, giving rise to a single, unilateral tumor, which is not hereditary. Hence, this type would occur without a family history and without any risk of passing the disease on to future generations.
Sporadic heritable
(30%). A sporadic mutation in the RB1 gene occurring in the sperm or the ovum forming the proband, or in in the early embryogenesis and involve his germ cells, so that one hit has already occurred and all cells of the body carry that mutation. Once the second hit occurs, the child develops retinoblastoma. The second hit may occur in many such predisposed cells, hence, these patients are more likely to develop the multifocal, and bilateral disease. This type would not have a family history, but there would be a risk of passing it on to future generations since it is a germline mutation.
Familial inherited
(10%). A family mutation has been inherited from the parent. All cells in the body carry this mutation. Hence, these patients are also likely to develop multifocal, and bilateral disease. Family history would be positive, as is the risk of passing on the disease to future generations. Often screening of parents may not show a typically malignant tumor, but a benign retinal tumor or retinocytoma. Sibling screening is important. Prenatal genetic testing is recommended, and in positive cases prenatal diagnosis of the disease is possible.
Moreover in bilateral sporadic and familial cases, RB1 gene mutation may produce other tumors like osteosarcoma, malignant melanoma, Ewing's sarcoma, etc.
Retinoblastoma - History
The name retinoblastoma was coined by Verhoff in 1926. As the name suggests, it is comprised of uncontrolled proliferation of immature cells (retinoblasts), of the inner layer of the eye; the retina. The possible hereditary nature of this disease was known since the early 1900s. Historically, the great physicist Newton had siblings (10 out of 16) lost to these diseases. However, times have changed and currently, there is greater than 90%survival of this once deadly disease.
Retinoblastoma - Epidemiology
Retinoblastoma has a global incidence of 1 in 15,000 live births. It is the commonest intraocular malignancy in our country. An estimated 2000 new cases are diagnosed in India every year. Almost 90% of cases present before the age of 3 years. The mean age of diagnosis is 18 months. In about 1/3rd of cases, the disease could be bilateral. Bilateral cases tend to present earlier (mean age of diagnosis s 14 months) versus unilateral (24 months) Sometimes, the tumor may start developing in utero as well.
Presentation
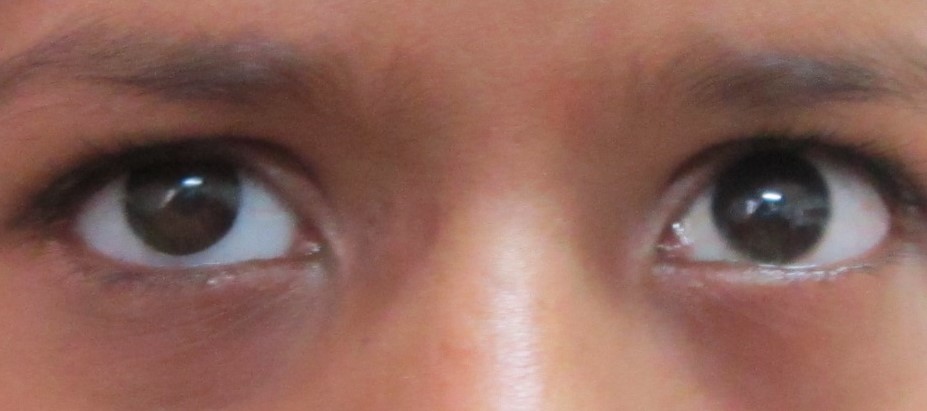
The most common presentation is leukocoria (white eye or cat's eye reflex). Other manifestations include poor visual acuity, squint, painful inflamed eye, proptosis, and retinal detachment.
Clinical Diagnosis
Retinoblastoma can be diagnosed clinically by examining the retina. It looks like a whitish mass arising from the retina with calcifications which can be verified by B scan ultrasonography. There may be associated with fluid, retinal detachment, and vitreous seeds.
Atypical presentations, or " masquerades " include acute uveitis and hypopyon, secondary glaucoma (increased eye pressure), chronic inflammation, and endophthalmitis with apparently no etiology, and painful proptosis resembling orbital cellulitis. A high index of suspicion and repeated examinations are vital to diagnosing such cases.
Investigations
Retinoblastoma is diagnosed by examination of the eye under general anesthesia using an ophthalmoscope. Calcifications and vitreous seedings confirm the diagnosis of retinoblastoma. An MRI or CT scan should be done to check for the intracranial extension of the disease. Routine bone marrow examination and CSF examination are not indicated except when suspicion of the spread of tumor beyond the globe e.g. patients with abnormal CBC or extension of tumor beyond the lamina cribrosa on ophthalmologic examination.
Retinoblastoma - Differential Diagnosis
A painless decreased vision with a white reflex must arouse suspicion of retinoblastoma. However, the closest differential is Coats's disease. This comprises of congenital retinal telangiectasias, that exudate fluid and cholesterol causing retinal detachment with a yellowish-white tumor-like appearance. Other differentials include cataract, retinopathy of prematurity, persistent hyperplastic primary vitreous, Toxocara granuloma, congenital coloboma, medullated nerve fibers, chronic endophthalmitis due to infection or trauma.
Treatment
The treatment options of retinoblastoma require a great deal of expertise in the response of the disease at its various stages, and the potentials of each treatment option. The treatment has undergone a paradigm of change over the past 50 years. From a near-fatal disease, it now has a 95% survival. The current management options target organ salvage and vision salvage. The current treatment guidelines are set as per the International Retinoblastoma Classification and TNM classification of the tumor and are followed uniformly across the globe.
- Chemotherapy: Systemic chemotherapy is now the mainstay and generally the 1st line of management. It aims at reducing the size of the tumors (chemo reduction) to allow for consolidation treatment with other agents to eradicate the tumors. Depending on the response to chemotherapy, other modalities are used in conjunction. Drugs used are Vincristine, Etoposide, carboplatin, cyclosporine.
- Sequentially aggressive local treatment (SALT):
This includes transpupillary thermotherapy (heating the Tumor), retinal lasers, cryotherapy (freezing the Tumor) and may be used alone in very small tumors or in conjunction with chemotherapy for larger ones. These technique targets organ as well as vision preservation.
Regional chemotherapy: Intravitreal chemotherapy (melphalan) may be given in selective cases. Periocular topotecan is another modality used. Recently, supra selective chemotherapy administered via the ophthalmic artery is being increasingly used across the centers. This technique avoids the systemic side effects of chemotherapy and avoids the removal of the eye as well. This technique has been aptly christened "chemosurgery”. Melphalan, carboplatin, and topotecan have been used for this technique
- Enucleation: Up to 25% -50% of the eyes may need eye removal or enucleation depending on the severity of the disease. Getting along the optic nerve stump (at least 15 mm) is very important, so is proper histopathology to confirm the tumor extent. A ball-like orbital implant is placed in the socket during primary surgery. A prosthetic eye can be fitted as early as 6 weeks post-operative with excellent outcomes. (FIGURE 3: Post enucleation prosthesis) Generally, these procedures don't interfere with any subsequent Tumor treatment modalities.
- Radiotherapy for retinoblastoma has a limited role in the current day scenario. It is generally resorted to in chemoresistant cases. It can be in the form of plaque brachytherapy for a unifocal medium-sized lesion or external beam radiation for multifocal disease with vitreous seeds.
Figure 3: Right eye ocular prosthesis after enucleation surgery and orbital implant
Introduction
Retinoblastoma is the most common primary tumor of the eye that arises from the nuclear layer of the retina and accounts for about 3% of the cancers occurring in children younger than 15 years of age.
Retinoblastoma can occur in both hereditary (40%) and sporadic (60%) forms. The hereditary disease includes those patients with positive family history and those patients who have inherited a germline mutation from an unaffected parent. It has been found associated with deletions or mutations of the "Retinoblastoma" gene on the q14band of chromosome 13. The germline form can manifest both as a unilateral or bilateral disease. However, most unilateral diseases are sporadic whereas all bilateral diseases are hereditary. But, unilateral disease in an infant is more likely to be a hereditary condition. 95% of the diagnosed cases of Retinoblastoma occur before the age of 5 years.
Presentation
The most common presentation is leukocoria (white eye or cat's eye reflex). Other manifestations include poor visual acuity, squint, painful inflamed eye, proptosis, and retinal detachment.
Investigations
Retinoblastoma is diagnosed by examination of the eye under general anesthesia using an ophthalmoscope. Calcifications and vitreous seedings confirm the diagnosis of retinoblastoma. An MRI or CT scan should be done to check for the intracranial extension of the disease. Routine bone marrow examination and CSF examination are not indicated except when suspicion of the spread of tumor beyond the globe e.g. patients with abnormal CBC or extension of tumor beyond the lamina cribrosa on ophthalmologic examination.
Tumor Staging
For the purpose of treatment, retinoblastoma is divided into the intraocular and extraocular disease.
Intraocular:
Tumor is localized to the eye, it does not extend beyond the eye or to other parts of the body.
Extraocular:
Tumor extends beyond the eye. It may be limited to the tissue around the eye, or it may spread to the CNS or other parts of the body.
Reese and Ellsworth have classified intraocular Retinoblastoma as follows:
Group 1
: very favorable for maintenance of sight.
- Solitary tumor, smaller than 4 disc diameters, all at or beyond the equator.
- Multiple tumors, none bigger than 4 disc diameters, all at or beyond the equator.
Group 2
: favorable for maintenance of sight.
- Solitary tumor, 4-10 disc diameter in size, at or beyond the equator.
- Multiple tumors, 4-10 disc diameter in size, behind the equator.
Group 3
: Possible for maintenance of sight.
- Any lesion anterior to the equator
- Solitary tumor larger than 10 disc diameter, posterior to the equator.
Group 4
Unfavorable for maintenance of sight
- Multiple tumors, some larger that 10 disc diameter in size
- Any lesion extending anterior to the ora-serrata.
Group 5
Very unfavorable for maintenance of sight
- Massive tumors involving more than one half of the retina.
- Vitreous seeding
(About 90% of patients present with Group5 disease in one or both the eyes)
Treatment
The goals of therapy are:
- To cure the disease
- To preserve as much sight as possible
I] TREATMENT FOR INTRAOCULAR RETINOBLASTOMA
The various options are:
- Enucleation: if tumor is large or useful vision is not expected.
- External beam radiation between 3500 to 4600 cGY, delivered in 17-25 fractions over 4-5 week period.
- Cryotherapy: For lesion smaller than 4-disc diameter in the anterior portion of retina, in addition to radiation or in place of photocoagulation.
- Laser Photocoagulation: For posteriorly located tumors smaller than 4-disc diameter distinct from the optic nerve head and macula.
- Systemic Chemotherapy: To reduce tumor load and avoid long-term effects of radiation therapy for patients with intraocular tumors not amenable to cryotherapy or photocoagulation alone.
Unilateral disease
- Enucleation: It is the standard therapy as most unilateral tumors are massive.
- Radiation, cryotherapy, photocoagulation, systemic chemotherapy are useful for smaller size tumors.
Bilateral disease
- Since one eye may be more severely involved as compared to the other eye, the standard treatment is to enucleate the more involved eye. However, if there is a potential for sight in both eyes, bilateral irradiation with close follow-up for response is indicated.
- Systemic chemotherapy followed by cryotherapy/photocoagulation is still experimental.
II] TREATMENT FOR EXTRAOCULAR RETINOBLASTOMA
There is no clear-cut therapy, however orbital irradiation with chemotherapy may be useful with/without intrathecal methotrexate.
Retinoblastoma - Prevention
All siblings of patients with Retinoblastoma should be examined periodically and DNA polymorphism analysis should be done.
Complications
Apart from the loss of vision and systemic spread of the disease, it has been found that 5% to 15% of children with either familial, multifocal, or bilateral Retinoblastoma may develop an intracranial neuroblastic tumor (pinealoma). Also, patients with germline type of tumor may have an increased risk of second malignant neoplasms e.g. osteosarcomas, soft tissue sarcomas & melanomas. Children with germline tumors may develop bilateral disease even if only one eye is involved at presentation. Hence, they should be examined frequently for the development of new Retinoblastoma tumors. It has been recommended that they should be examined every 2 to 4 months for at least 28 months and following treatment followed up until 7 years of age.
Retinoblastoma - Relapse
If the relapse is confined to the eye and the tumor is small, it can be treated with local therapy alone. The prognosis for vision and survival is excellent.
If a relapse is confined to the eye but is extensive, treatment with etoposide & carboplatin can be tired. The prognosis for vision is poor but survival remains excellent. If a relapse is extraocular, the prognosis is less than 50%.